


More than 47 million people are living with dementia worldwide and this number is predicted to increase to 131 million by 2050. Not only can dementia be a devastating condition, it carries a large economic burden with a worldwide cost estimated at US$818 billion (Prince Reference Prince, Comas-Herrera and Knapp2016). With no cure and an ageing population, the increasing prevalence is a worry.
Biomarkers are naturally occurring markers of the underlying pathological process of a particular disease. Numerous biomarkers to detect Alzheimer's disease have been developed over the past decade. These have helped to develop the theory that Alzheimer's disease is a continuum, which starts with the accumulation of Alzheimer's pathology years before the emergence of clinical symptoms. The continuum begins with a preclinical phase (Box 1), in which there are pathological changes of Alzheimer's disease (which can be detected by biomarkers), but no symptoms of dementia. This stage may pre-date Alzheimer's disease by decades (Jack Reference Jack, Knopman and Jagust2010). It is suggested that this progresses to a prodromal phase of mild symptoms that do not affect daily living. The final stage is established Alzheimer's dementia. Moving through these stages is not inevitable, and biomarkers have been developed to help predict who will show progression along the continuum (Dubois Reference Dubois, Feldman and Jacova2014).
BOX 1 Glossary of terms
Mild cognitive impairment (MCI)
Variously defined, but includes subjective memory symptoms or cognitive symptoms or both, objective memory impairment or cognitive impairment or both, and generally unaffected activities of daily living; affected people do not meet currently accepted diagnostic criteria for dementia or Alzheimer's disease
Amnestic mild cognitive impairment (aMCI)
A more specific term describing a subtype of mild cognitive impairment, in which there are subjective memory symptoms and objective memory impairment; other cognitive domains and activities of daily living are generally unaffected; affected people do not meet currently accepted diagnostic criteria for dementia or Alzheimer's disease
Preclinical Alzheimer's disease
The long asymptomatic period between the first brain lesions and the first appearance of symptoms and which concerns normal individuals who later fulfil diagnostic criteria for Alzheimer's disease
Prodromal Alzheimer's disease
The symptomatic predementia phase of Alzheimer's disease, generally included in the mild cognitive impairment category; this phase is characterised by symptoms not severe enough to meet currently accepted diagnostic criteria for Alzheimer's disease
(Dubois Reference Dubois, Feldman and Jacova2007)Preclinical Alzheimer's disease was initially a hypothetical model, but with the development of biomarkers, the in vivo pathological process can now be detected. The pathological hallmarks of Alzheimer's disease are amyloid plaques in the brain and hyperphosphorylated tau fibrillary tangles. Biomarkers of the disease therefore reflect the presumed underlying pathological processes (Livingston Reference Livingston, Sommerlad and Orgeta2017).
The detection of a preclinical phase of Alzheimer's disease is considered important for several reasons. First, it may contribute to our understanding of the pathogenesis of the disease itself. Second, this is the stage at which potential future treatments will be crucial, since it will allow disease-modifying drugs to be started before the onset of irreversible neurodegeneration (Berti Reference Berti, Polio and Lombardi2016).
A working group of the Research Institute of the Alzheimer's Association and the National Institute on Aging (Herskovits Reference Herskovits and Growden2010) has suggested that useful biomarkers in Alzheimer's disease would detect the underlying neuropathology of the disease with a sensitivity of >80% (this would be the true-positive rate) and a specificity of >80% (this would be the true-negative rate and would distinguish Alzheimer's disease from other types of dementia). In addition, the test should be affordable, reliable and non-invasive (De Meyer Reference De Meyer, Shapiro and Vanderstichele2010).
Box 1 helps to clarify the terminology used in this field. It will be noted, however, that the terms are not used exclusively: prodromal Alzheimer's disease seems to be a subset of mild cognitive impairment (MCI) and difficult to distinguish from amnestic MCI (aMCI). It is probably true that aMCI is better characterised than prodromal Alzheimer's disease, having been investigated as an entity for longer, albeit biomarkers might make these categorisations more precise. Of course, although aMCI is thought to be the type of MCI that is most likely to progress to Alzheimer's disease, there is no commitment to this in the name; whereas there is a commitment that prodromal Alzheimer's disease is the precursor of Alzheimer's disease. If this is not always the case, then ‘prodromal Alzheimer's disease’ is a misnomer and one that has the potential to cause harm. As we will suggest towards the end of this article, on this ground it would be unethical to label patients in this way if there is uncertainty.
The pathophysiology of Alzheimer's disease
The underlying pathophysiology of Alzheimer's disease is important in understanding why particular biomarkers have been developed and is depicted in Fig. 1. Alzheimer's disease involves abnormal functioning of amyloid precursor protein, which leads to an excess of amyloid-β in the cortex. This excess of amyloid-β is thought to lead to faulty accumulation of tau proteins, which in turn leads to synaptic dysfunction and neuronal death (Jack Reference Jack, Knopman and Jagust2010). The pathological hallmarks of Alzheimer's disease are amyloid-β plaques and neurofibrillary tangles of hyperphosphorylated tau. Compared to amyloid-β plaque formation, hyperphosphorylated tau and neurofibrillary tangles are more closely correlated to neurodegeneration and clinical symptoms (Bennett Reference Bennett, Schneider and Wilson2004).

FIG 1 A hypothetical model of the pathophysiological cascade in Alzheimer's disease (from Sperling Reference Sperling, Aisen and Beckett2011).
The difference between genetic profiles, other risk factors for dementia and cognitive reserve may explain the variation in lag time between the development of Alzheimer's pathology and dementia itself. Cognitive reserve is given as one explanation of why there is such individual variability in the time taken between developing Alzheimer's pathology and clinical symptoms (Fig. 2). The greater your cognitive reserve, the more insult you can endure before displaying cognitive symptoms. Cognitive reserve is an active process which can be strengthened by, for example, education and mental activity (Stern Reference Stern2012).
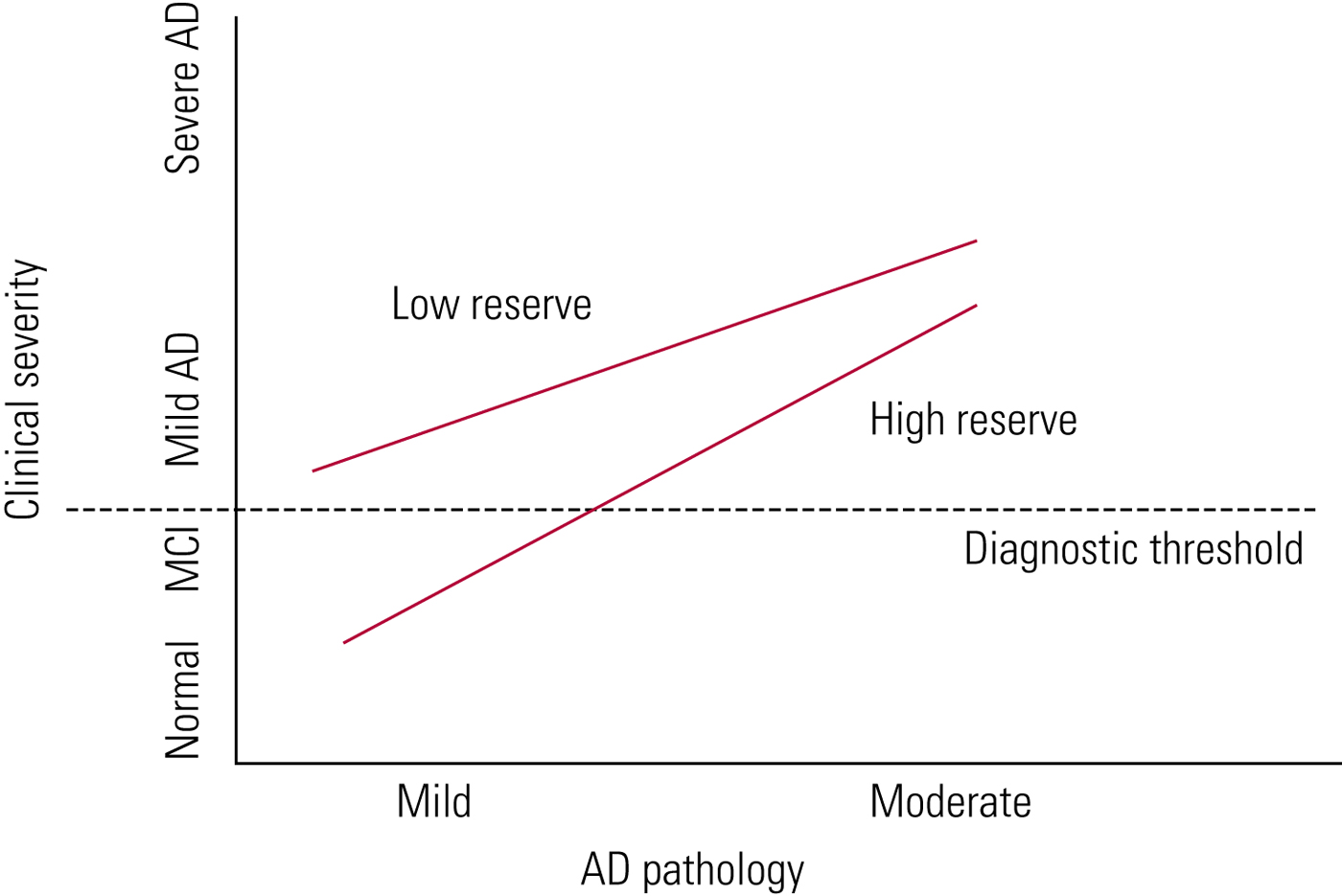
FIG 2 Cognitive reserve in ageing and Alzheimer's disease (AD) (Stern Reference Stern2012). With high cognitive reserve, more Alzheimer's disease pathology can be tolerated before symptoms develop.
Biomarkers
The main biomarkers of Alzheimer's disease can be divided by two major pathological processes: amyloid-β deposition and neurodegeneration. Hence, main biomarkers focus on:
• measures of brain amyloid-β deposition
• measures of markers of neurodegeneration.
The two main methods of detecting amyloid-β deposition are by measuring amyloid-β levels in the CSF and through position emission tomography (PET) amyloid imaging (Berti Reference Berti, Polio and Lombardi2016). Amyloid-β42 is the most likely type of amyloid to aggregate and is the most commonly measured amyloid variant. Where there is significant amyloid-β accumulation, levels of amyloid-β42 in the cerebrospinal fluid (CSF) are low and there is increased amyloid tracer retention on PET imaging (Sperling Reference Sperling, Aisen and Beckett2011).
Plasma amyloid can also be detected, but plasma amyloid-β42 has hitherto been used less frequently than CSF owing to its lower sensitivity and specificity (Anoop Reference Anoop, Singh and Jacob2010). Recently, however, there have been further advances in finding a suitable method to detect biomarkers in the blood (Nakamura Reference Nakamura, Kaneko and Villemagne2018). Acquiring blood, unlike CSF, requires a much less invasive procedure and, unlike PET scanning, is less costly. The researchers, by using immunoprecipitation and mass spectrometry, were able to achieve roughly 90% accuracy (using 11C-labelled Pittsburgh Compound B ([11C]PiB) PET as their gold standard) in the detection of amyloid-β biomarkers. They concluded: ‘These results demonstrate the potential clinical utility of plasma biomarkers in predicting brain amyloid-β burden at an individual level. These plasma biomarkers also have cost-benefit and scalability advantages over current techniques, potentially enabling broader clinical access and efficient population screening’ (Nakamura Reference Nakamura, Kaneko and Villemagne2018).
Neurodegeneration is measured by various indicators: increased concentrations of CSF total tau (t-tau) and phosphorylated tau (p-tau), hypermetabolism on fluorodeoxyglucose (FDG) PET imaging and atrophy in structural magnetic resonance imaging (MRI): t-tau is a more direct marker of neuronal degeneration and p-tau is a marker of neurofibrillary tangles (Dubois Reference Dubois and Albert2014).
Both CSF amyloid-β42 and tau protein have been found to reflect the degree of amyloid load and neurofibrillary abnormality accurately at autopsy (Tapiola Reference Tapiola, Alafuzoff and Herukka2009). Similarly, amyloid imaging strongly correlates with the pathological burden of disease at autopsy (Backsai Reference Bacskai, Frosch and Freeman2007) and with concentrations of amyloid-β in CSF (Fagan Reference Fagan, Mintum and Mach2006).
A significant number of cognitively healthy older people will have evidence of amyloid-β deposition both at autopsy and in biomarkers of CSF and PET amyloid imaging. The number of individuals who are biomarker ‘amyloid positive’ but ‘clinically negative’ varies from 20 to 40% (Sperling Reference Sperling, Aisen and Beckett2011), which is similar to autopsy findings (Morris Reference Morris, Storandt and McKeel1996). One theory is that if ‘amyloid-positive’ individuals lived longer they would eventually develop symptoms of Alzheimer's disease.
Jack et al (Reference Jack, Knopman and Jagust2010) have proposed a model to represent the progression of Alzheimer's pathology (Fig. 3). In this model, amyloid-β deposition occurs first, years before the onset of clinical symptoms. The duration of this phase may vary, depending on the individual's cognitive reserve and risk factors for Alzheimer's disease. Next, tau-mediated neurodegeneration begins, which is evident from changes on structural imaging. Finally, there is progression to cognitive impairment and clinical symptoms become evident. Important to the biomarker model is that Aβ accumulation alone is not sufficient to cause dementia (Jack Reference Jack, Knopman and Jagust2010).
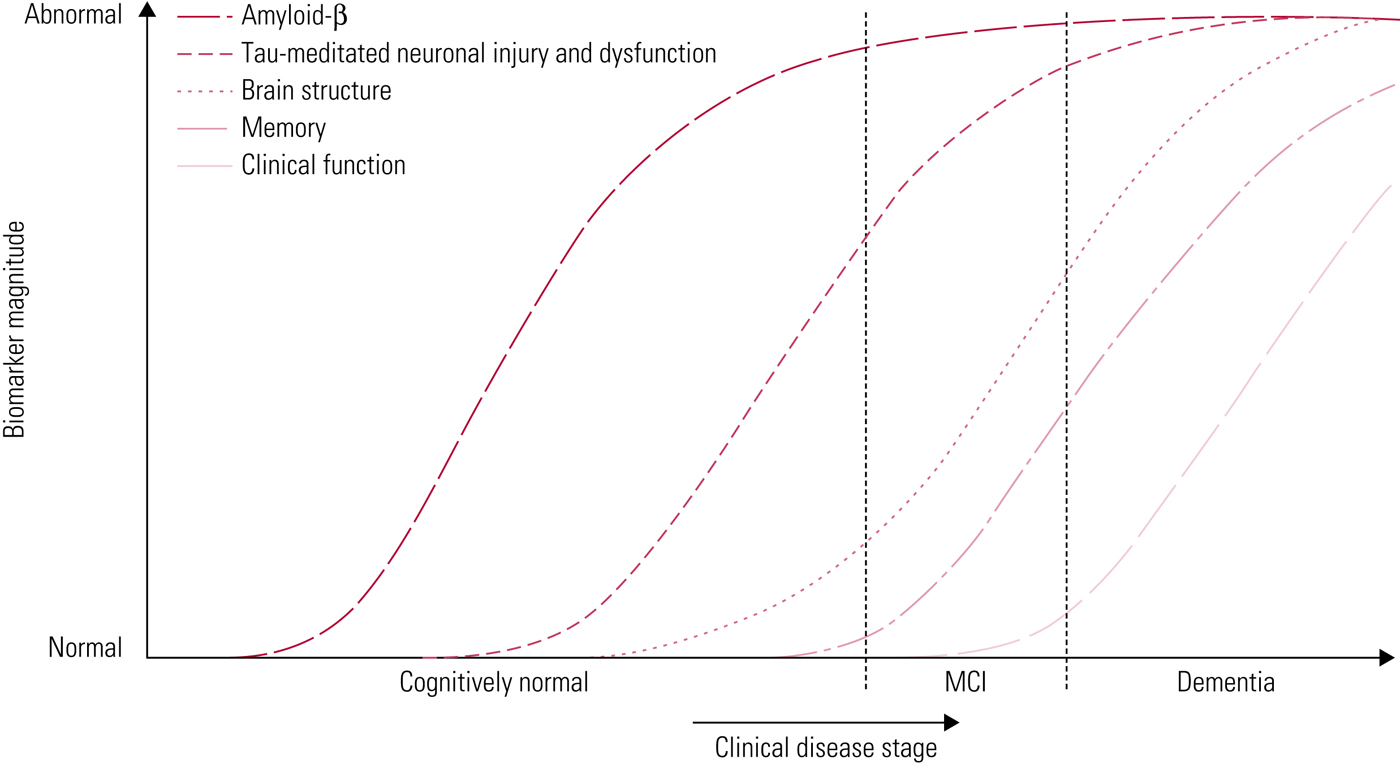
FIG 3 Accumulation of markers of disease over time (Jack Reference Jack, Knopman and Jagust2010). In this hypothetical model, amyloid-β markers are the first to become abnormal, followed by markers of neurodegeneration, followed by clinical symptoms. MCI, mild cognitive impairment.
Ocular biomarkers are less well-known biomarkers of Alzheimer's disease. Ocular biomarkers have been proposed for several reasons. First, individuals with Alzheimer's disease often present with visual deterioration, and this appears to pre-date cognitive changes. Second, the retina is easily visualised and ocular biomarkers do not require invasive tests such as lumbar puncture. There are many proposed ocular biomarkers, which we shall not discuss exhaustively here. For instance, micro-RNA can be found in tear fluid and is implicated in the regulation of amyloid. Other important approaches include detecting retinal amyloid-β accumulation, or an assessment of functional and clinical changes within the visual system (Lim Reference Lim, Li and He2016). Amyloid-β detection in the retina has been performed with or without contrast enhanced imaging. Further work is required to determine whether amyloid accumulation detected in the retina is reflective of brain deposition and, indeed, predictive of future cognitive impairment. Functional changes to the visual system include changes in neuronal responses, such as reduced visually evoked potentials over the occipital cortex and interrupted neurotransmission between photoreceptors in the retina. Further ocular biomarkers include ‘fixation and movement errors’, where there is failure to fixate or follow a target and reduced eye movements (Lim Reference Lim, Li and He2016).
Do CSF biomarkers accurately predict who will develop Alzheimer's dementia?
Using biomarkers to help predict who will develop dementia already has a significant history, particularly in determining which individuals with MCI are most at risk of developing Alzheimer's disease. In this population, there is evidence that biomarkers enhance diagnostic specificity and prognostication (Hardy Reference Hardy and Selkoe2002; Klein Reference Klein, Stine and Teplow2004; Hansson Reference Hansson, Zetterberg and Buchhave2006; Parnetti Reference Parnetti, Lanari and Silvestrelli2006; Mattsson Reference Mattsson, Zetterberg and Hansson2009; Visser Reference Visser, Verhey and Knol2009; Chetelat Reference Chetelat, Villemagne and Pike2011).
The combination of high CSF t-tau and p-tau with low CSF amyloid-β42 has been termed the ‘Alzheimer's disease signature’ (De Meyer Reference De Meyer, Shapiro and Vanderstichele2010) and is highly predictive of Alzheimer's dementia, as confirmed in three large multicentre studies: the Alzheimer's Disease Neuroimaging Initiative (ADNI) study, the Development of Screening Guidelines and Criteria for Predementia Alzheimer's Disease (DESCRIPA) study and the Swedish Brain Power (SBP) project. The CSF Alzheimer's disease signature has been shown to increase diagnostic accuracy significantly even at a prodromal stage, with a sensitivity of 90–95% and a specificity of about 90% for Alzheimer's disease (Snider Reference Snider, Fagan and Roe2009; De Souza Reference De Souza, Lamari and Belliard2011).
De Meyer e t al (Reference De Meyer, Shapiro and Vanderstichele2010) studied participants from the ADNI. The ADNI was established in 2004 to determine whether biomarkers could predict progression from MCI to Alzheimer's disease. Their sample included cognitively normal individuals, individuals with MCI and individuals with Alzheimer's disease. The Alzheimer's disease signature was detected in 90% of individuals with Alzheimer's disease, 72% of those with MCI and 36% of cognitively healthy individuals. In individuals with MCI and an Alzheimer's disease signature, the diagnostic sensitivity of progression to Alzheimer's disease was 90%, with a specificity of 64%. In addition, the combination simply of high CSF p-tau and low CSF amyloid-β42 correctly identified 100% of individuals with MCI who progressed to Alzheimer's disease.
Hansson et al (Reference Hansson, Zetterberg and Buchhave2006) found that the Alzheimer's disease signature had a sensitivity of >90% and specificity of >85% for individuals with MCI who subsequently developed Alzheimer's disease. Similarly, Van Rossum et al (Reference Van Rossum, Vos and Handels2010) found that the combination of CSF amyloid-β42 with either p-tau or t-tau was highly predictive of the development of Alzheimer's disease from MCI, with an odds ratio of 18.1 (95% CI 9.6–32.4).
Most of this evidence relates to individuals who already have MCI. If pathological changes really do start decades before the onset of clinical symptoms, is it possible to tell at this very early stage who will develop Alzheimer's disease in their later years? There have not been enough longitudinal studies to answer this conclusively, although some studies have found that amyloid-β positivity confers an increased risk of progression to Alzheimer's disease (Fagan Reference Fagan, Mintum and Mach2007; Li Reference Li, Sokal and Quinn2007; Villemagne Reference Villemagne, Pike and Darby2008; Morris Reference Morris, Roe and Grant2009; Storandt Reference Storandt, Mintun and Head2009; Resnick Reference Resnick, Sojkova and Zhou2010; Chetelat Reference Chetelat, Villemagne and Pike2011), and two (Vemuri Reference Vemuri, Wiste and Weigand2009; Yaffe Reference Yaffe, Weston and Graff-Radford2011) found that plasma biomarkers and imaging (MRI and FDG PET) can predict cognitively normal individuals who are at risk of cognitive decline. Fagan et al (Reference Fagan, Roe and Xiong2007) found that non-cognitively impaired individuals with the Alzheimer's disease signature progress to symptomatic cognitive impairment more quickly than do the remainder of the cohort.
Are CSF biomarkers useful in people who already have Alzheimer's disease?
A marked reduction in CSF amyloid-β42 has consistently been noted in patients at different stages of Alzheimer's disease. On the one hand, an isolated low amyloid-β42 is not sufficiently specific for a diagnosis. Non-Alzheimer's disease dementias, such as Lewy body disease and vascular dementia, are also associated with low CSF amyloid-β42 (Dubois Reference Dubois, Feldman and Jacova2014). CSF p-tau appears to be the most specific CSF biomarker, distinguishing Alzheimer's disease from non-Alzheimer's dementias (Koopman Reference Koopman, Le Bastard and Martin2009). On the other hand, combinations of CSF biomarkers appear to enhance the sensitivity of an Alzheimer's disease diagnosis, but there is no consensus as to which specific combination has the greatest accuracy (Dubois Reference Dubois, Feldman and Jacova2014). CSF biomarkers may also have a role in predicting the course of Alzheimer's disease. Snider et al (2009) found that individuals with Alzheimer's disease and the CSF Alzheimer's disease signature progressed more rapidly than those without. Markers of amyloid load tend to remain constant throughout the course of Alzheimer's disease, whereas t-tau and p-tau rise as Alzheimer's disease progresses (De Meyer Reference De Meyer, Shapiro and Vanderstichele2010).
Neuroimaging
Studies investigating the value of FDG PET (Box 2) in diagnosing prodromal Alzheimer's disease are few and have short follow-up periods. Metabolic reductions in the medial, temporal and parietal cortices, as well as the anterior and posterior cingulate, detected prodromal Alzheimer's disease with an accuracy of 75–84% (Dubois Reference Dubois, Feldman and Jacova2007). Differences between MCI and Alzheimer's disease have been consistently found on FDG PET (Shivamurthy Reference Shivamurthy, Tahari and Marcus2015).
BOX 2 FDG PET
Fludeoxyglucose (FDG) is an analogue of glucose and is used as a radiotracer in positron emission tomography (PET) imaging. FDG is taken up by cells and as a result acts as a marker of tissue metabolism. In Alzheimer's disease there are patterns of hypometabolism that are detected by poor FDG uptake. Different forms of dementia show distinct patterns of hypometabolism, which can aid diagnosis.
There are PET techniques that provide in-vivo detection of amyloid and, potentially, of neurofibrillary tangles. Imaging using [11C]PiB (N-methyl-[11C]2-(4'-methyl aminophenyl)-6-hydroxybenzothiazole) and [18F]FDDNP (2-(1-{6-[(2-[18F]fluoroethyl](methyl)amino]-2-naphthyl}ethylidene)malononitrile) have detected enhanced radio-ligand retention patterns in patients with Alzheimer's disease compared with controls; and positive [11C]PiB patterns are correlated with low CSF amyloid-β42 (Dubois Reference Dubois, Feldman and Jacova2007). There is evidence of enhanced [11C]PiB retention in cognitively normal people and in those with MCI (Dubois Reference Dubois, Feldman and Jacova2007). Longitudinal follow-up is required to determine whether these are people with a preclinical or prodromal dementia.
Diagnostic criteria
In 1984, a workgroup from the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS–ADRDA) (McKhann Reference McKhann, Drachman and Folstein1984) published criteria for the diagnosis of Alzheimer's disease. These criteria allowed only a ‘probable’ diagnosis of Alzheimer’s disease to be reached while the person was alive: a definitive diagnosis could be made only if Alzheimer’s pathology was found at autopsy. Furthermore, the (probable) diagnosis required the presence of significant disability and impact on daily living, thus excluding MCI (which was not recognised as such at the time). The specificity of these diagnostic criteria, in terms of distinguishing Alzheimer's disease from other types of dementia, was low (Dubois Reference Dubois, Feldman and Jacova2014).
In 2011, the National Institute on Aging and the Alzheimer's Association (NIA–AA) published guidelines (Jack Reference Jack, Albert and Knopman2011) revising the 1984 diagnostic criteria. All-cause dementia continues to be diagnosed by history and neuropsychological testing, with impairment in function required for a diagnosis to be made. This is then further subdivided into ‘probable’ or ‘possible’ Alzheimer's disease, and biomarkers may be used as supportive information to help guide a diagnosis and to help distinguish Alzheimer's disease from other forms of dementia. There is also a shift away from concentrating only on memory problems as the clinical phenotype of Alzheimer's disease towards also including impairments of other cognitive domains, such as language (Dubois Reference Dubois, Feldman and Jacova2014).
The NIA–AA guidelines of 2011 also recognise the spectrum of Alzheimer's disease, such as preclinical Alzheimer's disease and MCI, and the use of CSF and imaging biomarkers in identifying these states. Dubois & Albert (Reference Dubois and Albert2004) describe prodromal MCI as a heterogeneous state that may or may not lead to dementia of multiple different aetiologies. They describe prodromal Alzheimer's disease as ‘MCI of the Alzheimer type’ and advocate the use of CSF and imaging biomarkers to help make this distinction. For preclinical Alzheimer's disease, however, the NIA–AA guidelines only apply in a research setting (Watkin Reference Watkin, Sikdar and Majurndar2013). Research criteria for diagnosing preclinical states of Alzheimer's disease developed by the International Working Group (the IGW-2 criteria) require the individual to be asymptomatic, have a marker of Alzheimer's disease pathology or an Alzheimer's disease autosomal dominant mutation on chromosome 1, 14 or 21 (Dubois Reference Dubois, Feldman and Jacova2014). This is describing an at-risk state where progression to Alzheimer's disease is not inevitable.
Limitations in the use of biomarkers
Biomarker results are often taken from highly selected populations, so that the predictability of the results is likely to be exaggerated (Livingston Reference Livingston, Sommerlad and Orgeta2017). A major limitation is that participants in these studies are generally volunteers and one wonders who volunteers for invasive trials such as these. They may be individuals with concerns about their cognitive performance or with a family history of dementia. This is supported by the higher rates of apolipoprotein E (ApoE) ε4 carriers detected in many of these cohorts (Sperling Reference Sperling, Aisen and Beckett2011). This would lead to an overly high prediction of biomarkers in the general population.
Lumbar punctures are not without risk and there is no standardised analytic technique or reference range for CSF biomarkers (Livingston Reference Livingston, Sommerlad and Orgeta2017). CSF biomarkers show 20–30% inter-laboratory and inter-assay variability (Berti Reference Berti, Polio and Lombardi2016). The Alzheimer's Association has funded the Alzheimer's Association QC [Quality Control] Program for CSF Biomarkers to try to reduce this variability by using reference laboratories to compare samples (Mattsson Reference Mattsson, Andreasson and Persson2013).
Ethical implications
With this new concept of preclinical dementia come numerous ethical implications. Finding out that a cognitively healthy individual has Alzheimer's pathology leaves the researcher with a difficult question regarding an unknown risk that the participant will develop an incredibly debilitating disease. With such a significant number (20–40%) of cognitively normal individuals with evidence of amyloid-β plaques, more information is needed on the sensitivity of such a test, so that the implication of this result can be communicated to participants. Currently, establishing preclinical dementia has no clinical utility, and an unknown prognosis therefore provides unsettling and uncertain information for the patient (Harkins Reference Harkins, Sankar and Sperling2015).
Hughes et al (Reference Hughes, Ingram and Jarvis2017) carried out a scoping review to explore some of the issues that arise following detection of preclinical dementia and identified four main themes: stigma, ethical questions, psychological burden and language/terminology.
First, individuals with preclinical dementia may face stigma, including difficulties at work and insurance implications. In addition, social stigma can lead to isolation and cause the individual to form negative self-perceptions (Hughes Reference Hughes, Ingram and Jarvis2017).
Second, the idea of non-maleficence has recurred in the literature. Harm may be done by telling someone about biomarker positivity, particularly if, as is currently the case, there are no effective treatments for Alzheimer's disease. However, the Risk Evaluation and Education for Alzheimer's Disease (REVEAL) project reported that participants were no more likely to develop depression, anxiety or distress on being told their APoE genotype in addition to general risk factors for Alzheimer's disease (such as family history, age and gender) than those who were told the general risk factors alone (Green Reference Green, Roberts and Cupples2009). In terms of beneficence, individuals with a positive amyloid profile can alter their lifestyle choices to reduce their risk of dementia in the future or have time to make advance care planning decisions.
Third, there are psychological burdens associated with these studies, including anxiety about the test results. Psychoeducation to help participants and their families understand and reflect on the results may help.
The final theme in the literature is the importance of language. How things are communicated will shape how the person understands the meaning of results that may be ambiguous. Indeed, language itself can cause harm. A question can be raised, given the level of uncertainty, about whether the label of ‘preclinical Alzheimer's disease’ should ever be used, except perhaps in the context of research. The situation described by ‘preclinical Alzheimer's disease’ can also be called ‘non-dementia Alzheimer's disease’. It could be said that those affected are not ill, but have a disease. This is not uncommon in medicine: for example, many people may have cancer but no symptoms. It is more problematic when people have the putative pathology, but never go on to have symptoms. So, for example, many normal older people will have amyloids in their brains and it seems odd on this account alone to describe them as diseased. The worry here is a broad one about the medicalisation of ageing (Estes Reference Estes and Binney1989).
Conclusions
At present, CSF biomarkers are not radically altering the diagnostic pathway for Alzheimer's disease. The core criteria for diagnosing Alzheimer's disease remain a gradual, progressive cognitive impairment that is documented on objective testing. History and examination, therefore, remain the crucial elements of diagnosis, which can be supported by the use of in vivo markers of Alzheimer's disease pathology. CSF and imaging biomarkers are providing more information, which will need to be applied in light of the individual presentation, and diagnostic criteria may continue to change as we learn more about their diagnostic accuracy (Dubois Reference Dubois, Feldman and Jacova2014). In MCI and in established dementia, biomarkers do appear to provide useful diagnostic and prognostic information.
According to the working group for the Alzheimer's Association and the National Institute on Aging, the CSF Alzheimer's disease signature is coming close to fulfilling the desirable features of a useful biomarker (De Meyer 2010; Herskovits Reference Herskovits and Growden2010). What about using Alzheimer's disease biomarkers as a screening tool (Box 3)? According to Wilson's criteria for screening (Wilson Reference Wilson and Jungner1968), Alzheimer's disease is an important health problem with a recognisable pathophysiological process. There is an early symptomatic stage (prodromal Alzheimer's disease) and there are tests which are (arguably) easy and acceptable to perform. The tests, however, are not sensitive or specific enough and there currently is not an effective treatment for Alzheimer's disease. A sensible consensus, therefore, would be that these biomarkers are not appropriate for screening the general population and should be reserved for detecting preclinical dementia in research settings only.
BOX 3 Further questions
Should we be using CSF biomarkers in routine clinical practice?
This is a controversial question and the answer varies between different countries. In Scandinavia, CSF biomarkers are part of a routine work-up to diagnose Alzheimer's disease.
How invasive and safe are lumbar punctures?
The threshold varies between individuals. They are certainly more invasive than blood tests and they are associated with adverse effects, including pain, infection and headaches. Serious side effects, however, are very rare (Herskovits Reference Herskovits and Growden2010).
How would using CSF biomarkers work practically?
In the UK, patients with memory impairment are not necessarily reviewed by a neurologist. Psychiatrists are not trained to perform lumbar punctures so they would have to refer to a neurological (or other appropriate) team; this service design will require thought and may depend on local services and relationships.
What to do with the results?
In MCI, there is evidence that CSF biomarkers help to determine who will develop full-blown Alzheimer's disease. Similarly, in established Alzheimer's disease, CSF biomarkers may help in estimating the rate of progression or provide supportive evidence for a diagnosis to be made. However, in those who are cognitively normal, it will be more difficult to interpret the results. The results of studies to date are likely be overly optimistic, as the participants may have volunteered because of concerns regarding their memory or a positive family history of Alzheimer's disease. More longitudinal studies are required further to clarify sensitivities and specificities to establish whether these biomarkers will be useful in the cognitively normal.
MCQs
Select the single best option for each question stem
1 As regards mild cognitive impairment (MCI):
a it is also described by the clinical state prodromal dementia
b activities of daily living are affected
c in amnestic MCI other cognitive domains are affected before memory
d it is more likely to develop in those with stressful, high-powered careers
e it is a research rather than a clinical diagnosis.
2 In preclinical Alzheimer's disease:
a amyloid-β accumulation is sufficient to cause Alzheimer's disease
b tau neurodegeneration is the first pathophysiological process to occur
c the duration of this phase is always over a decade
d the duration of this phase before the development of clinical symptoms is affected by cognitive reserve
e this is a symptomatic phase before the development of Alzheimer's disease.
3 With CSF biomarkers:
a the ‘Alzheimer's disease signature’ is a combination of high CSF amyloid-β42 and high CSF p-tau
b the ‘Alzheimer's disease signature’ has been found in 72% of individuals with MCI
c the number of people who are ‘amyloid positive’ and ‘clinically negative’ varies between 40 and 50%
d the ‘Alzheimer's disease signature’ has a sensitivity >95% for progression to Alzheimer's disease in MCI
e Lewy body dementia is associated with a raised CSF amyloid-β42.
4 With regard to other biomarkers:
a ocular biomarkers include increased visually evoked potentials over the occipital cortex
b retinal amyloid-β accumulation can only be detected with contrast enhanced imaging
c amyloid-β deposition can be detected by position emission tomography (PET)
d FDG PET can detect MCI with an accuracy of 62–74%
e there are no imaging changes that can be detected in preclinical Alzheimer's disease.
5 With the NIA-AA diagnostic guidelines for Alzheimer's disease:
a function must always be impaired for a diagnosis of Alzheimer's disease
b at least three cognitive domains must be impaired
c biomarker status should always be used to make a diagnosis of Alzheimer's disease
d collateral information is no longer ever necessary to make a diagnosis of Alzheimer's disease
e abnormalities in the visuospatial domain is required for a diagnosis of Alzheimer's disease.
MCQ answers
1 a 2 d 3 b 4 c 5 a




eLetters
No eLetters have been published for this article.