



1. Introduction
Until about the 1950s, carbonatites were considered little more than a petrological curiosity, but in recent years there has been a resurgence of interest in their mineralogy and petrology. This is partly because of the realization that carbonate liquids might play a significant role in metasomatising the mantle, and by the appreciation that carbonatite-bearing complexes are the principal sources of niobium, as well being the predominant hosts of economically recoverable rare earth deposits.
Initially carbonatites were not accepted as igneous rocks regardless of the advocacy of such eminent petrologists as Högbom, Brögger and von Eckermann. The idea that a rock composed essentially of calcite or dolomite could have crystallized from magma, because of the extremely high melting temperature of calcite, was scoffed at even by such a luminary as Norman Bowen. However, with the groundbreaking discovery by Wyllie and Tuttle (Reference Wyllie and Tuttle1960) that calcite can melt in the presence of water, even in amounts unlikely to be present in the Earth’s mantle, the igneous origin came to be accepted. Subsequently, papers on carbonate-alkaline rock complexes now routinely list the following as possible origins of carbonatite-forming magmas:
-
(1) Direct melting of metasomatized mantle peridotite or ‘basaltic’ eclogite;
-
(2) Differentiation of a carbonate-bearing alkaline silicate magma produced by mantle melting;
-
(3) Separation by liquid immiscibility of carbonatite and silicate liquids from a precursor carbonate-bearing alkaline silicate magma such as nephelinite or melilitite;
-
(4) Melting of eclogite formed by metamorphism of subducted oceanic or continental crust.
Note that we refer in the title of this polemic to ‘carbonatite-forming magmas’ rather than the commonly used ‘carbonatite magma’ or ‘carbonatite melt’, as the actual composition of any calcite or dolomite carbonatite-forming magma is not known. Typically, many carbonatites appear to be rocks formed by crystal fractionation from a variety of uncharacterized parental carbonated silicate magmas and they certainly do not represent former liquid compositions. Unlike common magma types such as basaltic magmas, which crystallize to basalts or gabbros, there are no analogues with respect to carbonatites. The nyerereite–gregoryite lavas of Oldoinyo Lengai are unique and suggestions that these are parental to common calcite or dolomite carbonatites (Le Bas, Reference Le Bas, Fitton and Upton1987) have been rigorously invalidated by Twyman and Gittins (Reference Twyman, Gittins, Fitton and Upton1987). These objections remain regardless of recent proposals attempting to devise unified petrogenetic schemes linking calcite carbonatites and the Oldoinyo Lengai lavas (Weidendorfer et al. Reference Weidendorfer, Schmidt and Mattsson2017; Chayka et al. Reference Chayka, Kamenetsky, Vladykin, Kontonikas-Charos, Prokopyev, Stepanov and Krasheninnikov2021).
By 2023, liquid immiscibility has become overwhelmingly the preferred mode of magma genesis for the majority of those who investigate carbonatites. It is with this particularly frequent claim to its being the dominant factor in their derivation with which we are most concerned. We suggest that liquid immiscibility has become a convenient if unwarranted crutch on which to lean; one on which much ill-thought and erroneous opinion has been expended. A stage has been reached whereby ‘proof’ is merely a chain of citations of previous investigations where no reasonable case for liquid immiscibility was originally presented.
The objective of this contribution is to assess critically the proposed role of liquid immiscibility in the genesis of calcite and dolomite carbonatites. At the outset, we must state that we do not object to the concept of liquid immiscibility as a legitimate petrogenetic process. We recognize its well-documented role in certain aspects of basalt evolution (Roedder, Reference Roedder and Yoder1979; Zhang et al. Reference Zhang, Namur and Charlier2023) and formation of Cu–Ni–sulphide deposits (Naldrett, Reference Naldrett2004). We further note that although liquid immiscibility might play a significant role in the genesis of the unique Oldoinyo Lengai nyerereite-gregoryite lavas (Mitchell, Reference Mitchell1997, Reference Mitchell2009), extrapolation of hypotheses for their petrogenesis cannot, and should not, be extended to common alkaline-rock carbonatite complexes. Our principal objection to the current infatuation with liquid immiscibility is that adherents typically invoke the process without presenting any realistic geological evidence for its occurrence, together with uncritical acceptance of earlier laboratory experimental studies.
Are carbonate and silicate liquids by their nature immiscible? We suggest that they must be as a consequence of their physical properties (Jones et al. Reference Jones, Genge and Carmody2013), but that in a geological context, they are NOT conjugate. We consider the presumption that they are conjugate has had a stultifying effect on hypotheses of carbonatite genesis. The preoccupation with immiscibility interpretations of carbonatite petrogenesis is evident from a perusal of textbooks and papers over the past forty years as illustrated by the following examples:
‘There is now a consensus that their parental magma originates by the separation of an immiscible liquid from a CO2 saturated nephelinite or phonolite magma’ (Fitton & Upton, Reference Fitton and Upton1987, p. xiii).
‘Liquid immiscibility is probably the only magmatic process known which can explain the association of contemporaneous and discrete intrusions of carbonatites and alkaline silicate rocks’ (Le Bas, Reference Le Bas and Bell1989, p. 432).
‘Immiscibility can account for the many types of carbonatites more readily than any other process’ (Kjarsgaard & Hamilton, Reference Kjarsgaard, Hamilton and Bell1989, p. 403).
‘Liquid immiscibility has been widely recognized as one of the underlying processes which generates carbonatite magma from a silica undersaturated parent magma at crustal depths’ (Ray, Reference Ray1998, p. 3301).
‘However, there is increasing consensus that many carbonatites form as immiscible melts within alkaline silicate magmatic systems’ (Goodenough et al. Reference Goodenough, Deady, Beard, Broom-Fendley, Elliot, van den Berg and Öztürk2021, p. 19).
‘Silicate-carbonatite immiscibility is a crucial process that precedes the crystallization of most carbonatites on the planet’ (Berkesi et al. Reference Berkesi, Myovela, Yaxley and Guzmics2023, p. 42).
Liquid immiscibility has a long and chequered history in igneous petrology beginning with Norman Bowen’s dismissive statement in his epic book The Evolution of the Igneous Rocks, i.e. ‘It is usually merely stated that the original magma split into this magma and that magma. Apparently the authors of such statements do not realize that they have not in any way described or discussed a process but have merely restated, with a maximum of indirection, the observational fact that this rock and that rock are associated in the described field’. (p. 7; Bowen, Reference Bowen1928). Although this dismissal was somewhat moderated in the review by Roedder (Reference Roedder and Yoder1979) of evidence for liquid immiscibility in some lunar basalts and other mafic silicate rocks, we still consider it an accurate description of the common invocation of liquid immiscibility in discussions of carbonatite-forming magma genesis. While we accept that experimental studies have established that carbonate and silicate liquids are mutually immiscible, we do not believe that any studies have actually shown them to be conjugate. We begin our review with consideration of the experimental studies of carbonatite-related systems followed by a consideration of the geological evidence. The arguments usually proffered by supporters of liquid immiscibility can be summarized as follows:
-
(1) Experimental data demonstrate that nephelinite and phonolite magmas can produce separate silicate and carbonate liquids by liquid immiscibility.
-
(2) The very rare presence in some alkaline silicate rocks of calcite ocelli, which are considered analogous to calcite of similar morphology, formed in many experimental studies and interpreted as quenched drops of pure calcite liquid.
-
(3) Bulk rock trace element contents of the silicate and carbonate rocks are consistent with experimentally derived partition coefficients between co-existing silicate and carbonate liquids.
-
(4) Radiogenic isotope ratios of silicate and carbonate rocks are similar.
-
(5) Bimodal discrete suites of silicate and carbonate rocks commonly occur in close proximity within an igneous complex and usually without any apparent significant difference in time of emplacement.
-
(6) In calcite carbonatites, the magnesium content is low and thus these rocks cannot have crystallized from a mantle-derived magma because such magma must have high magnesium contents; hence this favours liquid immiscibility.
A common reason for adopting the concept of liquid immiscibility to explain the origin of carbonatite-forming magma is that it is seen as the only solution to a problem, which is itself wholly imagined. Thus. it is reasoned that most carbonatites are calcitic and since rock composition directly reflects the parent magma composition, they must have formed from a calcitic magma. As magmas derived from direct melting of mantle peridotite are dolomitic, they cannot precipitate calcite. Therefore, the parent magma of calcitic carbonatites cannot have originated through direct mantle melting, and another source must be found. This leaves the remaining possibilities of progressive differentiation of a nephelinitic magma enriched in CO2, or a similar magma undergoing liquid immiscibility. Many studies opt for liquid immiscibility because it has supposedly been demonstrated experimentally by Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989) and Kjarsgaard (Reference Kjarsgaard1998), thus ignoring all possibilities of fractional crystallization. However, the reasoning is rife with false assumptions and Gittins and Harmer (Reference Gittins and Harmer2003) have even suggested that it might be called Desperation Petrology for it is well established experimentally that a dolomite magma can crystallize calcite; a topic which is discussed further in section 11 of this work.
2. Experimental discovery of liquid immiscibility in carbonatite-related systems
In the mid-1950s, Frank Tuttle’s initiative led to the discovery by Wyllie and Tuttle (Reference Wyllie and Tuttle1960) that calcite can crystallize as a liquidus phase at temperatures as low as 650oC at 0.1 GPa, in the presence of water; albeit in very large amounts. As a consequence of this seminal work, doubts about carbonatites being magmatic rocks were essentially assuaged.
Subsequently, numerous experimental studies were initiated with the investigation of calcite-containing synthetic systems whose compositions were far removed from the composition of magmas such as nephelinites or melilitites, which have been proposed as parental to calcite and/or dolomite carbonatites. In the majority of these studies, immiscibility was observed between silicate and calcitic liquids. However, a major problem in the experimental design of all these initially investigated systems, discussed in more detail below, is that the selection of components inevitably ensured that the carbonate liquids produced would be calcitic or sodium-rich.
As early as Reference von Eckermann1948, von Eckermann proposed that the primary carbonatite magma of the Alnö complex (Sweden) was alkali-rich. This hypothesis promoted several investigations of the system NaAlSi3O8–Na2CO3–CO2 up to 0.25 GPa by Kooster van Groos and Wyllie (Reference Kooster van Groos and Wyllie1963, Reference Kooster van Groos and Wyllie1966, Reference Kooster van Groos and Wyllie1968, Reference Kooster van Groos and Wyllie1973) to assess its viability. In these systems, it was found that there is a wide field of immiscibility in which an alkali-rich carbonate liquid is separated from an albite-rich silicate liquid, which the addition of water did not suppress. It was found that the immiscibility gap did not reach alkali-free compositions and that conjugate carbonate liquids contained significant Na2CO3 (∼10 wt.%) in addition to silicate components. We consider that, although of interest to studies of phase equilibria in plagioclase-bearing systems, they are of dubious value in explaining the origins of the common Na-poor calcite carbonatites. However, it was suggested that these data might be relevant to the genesis of the then recently discovered nyerereite–gregoryite lavas erupted by the Tanzanian volcano Oldoinyo Lengai.
Subsequent study of synthetic systems [(SiO2–Na2O–Al2O3–CaO)+CO2±H2O; albite-calcite; nepheline–albite–calcite] by Watkinson and Wyllie (Reference Watkinson and Wyllie1971); Kjarsgaard and Hamilton (1988, Reference Kjarsgaard, Hamilton and Bell1989); Brooker and Hamilton (Reference Brooker and Hamilton1990); Lee et al. (Reference Lee, Wyllie and Rossman1994); Lee and Wyllie (Reference Lee and Wyllie1994); Marakushev and Suk (Reference Marakushev and Suk1998) and Suk (Reference Suk2001) elaborated on the earlier work. A summary of much of these data can be found in Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989). The investigations presented many contrary results and interpretations of the experimental data arising primarily as a consequence of different experimental conditions and bulk compositions.
The studies of Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988) are of particular importance in the investigations of liquid immiscibility in synthetic systems as it is purported that they established the presence of a pure CaCO3 melt in the form of rounded and dumbbell-shaped calcite crystals in silicate glass (albite or anorthite) – calcite systems. These studies were at temperatures from 1100–1250oC and a pressure of 0.5 GPa using albite glass-calcite mixtures (Ab15CC85 to Ab75CC25: wt.%), with, and without, the addition of up to 37 wt.% Na2CO3. The authors claimed that at these temperatures and pressures, and for all alkali-bearing mixtures, there was liquid immiscibility between silicate and carbonate liquids. Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989) extended this study using similar albite glass-calcite mixtures at temperatures of 1250oC with pressures of 0.2 to 0.5 GPa and with one experiment (RB7: Ab38CC32) also at 1.5 GPa and 1300oC and showed that a similar two-liquid field exists at 0.2GPa and 1250OC (Fig. 1). These experiments were interpreted to define a silicate liquid limb and a Na–Ca–carbonate limb of a symmetrical solvus in the quaternary system [(SiO2+Al2O3)-CaO-Na2O-CO2]. Importantly, none of these experiments defined the consolute temperature of the proposed silicate–carbonate solvus as no supra-solvus experiments were undertaken. It is notable that all carbonate compositions contained up to 2 wt.% (SiO2+Al2O3), which might originate from the presence of silicate micro-inclusions as noted by Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988), and result in the carbonate limb of the solvus lying very close to, but not directly on the Na2O–CaO join. The principal evidence for liquid immiscibility presented by Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988, Reference Kjarsgaard, Hamilton and Bell1989) is that the silicate glass composition is not the same as that of the starting material and that textural features of the run-products are consistent with a pair of immiscible liquids in which the calcite is in the form of spheres in silicate glass; a texture which they interpreted as incontrovertible evidence of carbonate liquid immiscibility. We discuss this feature further in section 5. Two of their experimental runs (RB7 and BK10), on the basis of this texture, appear to indicate silicate liquids co-existing with pure CaCO3 ‘liquid’ which lead them to state: ‘Consequently, a pure CaCO 3 melt is immiscible with a silicate melt’ (Kjarsgaard & Hamilton, Reference Kjarsgaard, Hamilton and Bell1989, p. 391). However, this conclusion is simply impossible as it was at variance with the conditions of calcite melting known even at that time.

Figure 1. The ‘Hamilton Projection’ of Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989) experimentally determined silicate–carbonate liquid immiscibility solvus at 5 kb and 1250oC. Tie lines between co-existing liquids for different starting bulk compositions in the system albite–calcite–sodium carbonate are denoted by crosses. The tie line for the bulk composition RB7 (albite calcite) is also shown. Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988, Reference Kjarsgaard, Hamilton and Bell1989) have claimed that the round calcite formed in experiments BK10 and RB7 represents liquid calcite. Diagram after Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989).
All of these studies established the existence of silicate–carbonate immiscibility with a range of synthetic alkali-bearing carbonate liquid compositions, but they have not provided any satisfactory explanations for the genesis of calcite or dolomite carbonatites, although they might have relevance to Oldoinyo Lengai lavas. There were no investigations of dolomite-bearing systems at low pressure and only one, NaFe3+Si2O6-CaCO3, which included iron (Verwoerd, Reference Verwoerd1978). Although there is evidence of carbonatite-forming magmas at temperatures as high as 1000–1100oC (Gittins, Reference Gittins1978, Reference Gittins1979; Gittins et al. Reference Gittins, Harmer and Barker2005), the experiments at 1100–1300oC discussed above seem inordinately high for realistic application to carbonatite magmas genesis.
Lee and Wyllie (Reference Lee and Wyllie1994) concluded that alkali-enriched carbonatite melts capable of precipitating cumulate calcite could be generated in several ways, including fractional crystallization, as suggested by data from Watkinson and Wyllie (Reference Watkinson and Wyllie1971), but that there was no experimental evidence for petrological processes forming pure CaCO3 liquids at feasible temperatures and pressures. They proposed that the existence of ‘calciocarbonatite magma’ requires substantiation by a viable process.
Regardless of the caveat of Lee and Wyllie (Reference Lee and Wyllie1994), much of the current popularity of liquid immiscibility as a petrogenetic process derives from the study of these synthetic systems. Without these discoveries there is little likelihood that the concept of carbonatite-forming magma genesis by liquid immiscibility would have emerged. However enticing it might be to extrapolate these studies to carbonatite genesis sensu lato, it is insufficient to do so without supporting evidence from naturally occurring rocks. Thus, given the limitations of experiments using synthetic systems, lacking many of the major elements occurring in carbonatites, attention turned to investigations using natural rocks. It is fundamentally important to note that no experiments have produced an immiscible dolomite liquid, thus eliminating liquid immiscibility as a possible explanation for the genesis of numerous dolomitic carbonatites.
3. Experiments with naturally occurring rocks rather than synthetic systems
Attempts to relate experiments to geological processes were significantly influenced initially by the discovery of the nyerereite–gregoryite lavas, commonly called natrocarbonatite, erupted by the volcano Oldoinyo Lengai (Dawson, Reference Dawson1962), and by hypotheses suggesting calcite carbonatites could be derived from magmas of these compositions (Le Bas, Reference Le Bas, Fitton and Upton1987). The investigations began with those of Freestone (Reference Freestone1978), followed by Hamilton et al. (Reference Hamilton, Freestone, Dawson and Donaldson1979) and Freestone and Hamilton (Reference Freestone and Hamilton1980), which all showed that molten nephelinite and phonolite silicate lavas from Oldoinyo Lengai, or melted synthetic calciocarbonatite and natrocarbonatite are immiscible in the pressure range 0.07–0.76 GPa and temperatures from 900 to 1250oC. However, none of these experiments were undertaken at a sufficiently high temperature to create initially a single-phase liquid that, upon cooling, exsolved to produce two immiscible liquids. All that was shown is that the two liquids are immiscible at the temperatures and pressures of the studies; it does not demonstrate that they are conjugate. The compositions of the silicate and carbonate liquids obtained in the experiments were presented in a ternary [(SiO2+Al2O3)-Na2O-CaO)] diagram, which has become known as the Hamilton projection (Fig. 1). The data projections illustrated in fig. 2 of Hamilton et al. (Reference Hamilton, Freestone, Dawson and Donaldson1979) and figs. 3 and 5 of Freestone and Hamilton (Reference Freestone and Hamilton1980) show that none of the experiments actually defined the shape of any potential solvus, or the closure composition and temperature of the solvus, which was assumed to be symmetrical. In the Hamilton et al. (Reference Hamilton, Freestone, Dawson and Donaldson1979) experiments, equal weights of silicate and carbonates were used as starting compositions for the experiments, and subsequent experiments by Freestone and Hamilton (Reference Freestone and Hamilton1980) used from 15 to 80 wt.% of carbonate components. Such extremely large amounts of carbonates resulted in bulk compositions, which have little or no resemblance to any naturally occurring nephelinite or phonolite. They simply exceed the solubility of carbonates in silicate liquids so virtually guaranteeing the production of immiscible liquids in the experiments, given that silicate and carbonate liquids are intrinsically immiscible. For such experiments, liquid immiscibility is a pre-ordained event and certainly they do not prove that nephelinite or phonolite can be parental even to ‘natrocarbonatite’ magma.
Importantly, it is commonly forgotten that these experiments were concerned essentially with producing ‘natrocarbonatite’ liquids and hence have little application to calcite and dolomite carbonatites. Lee and Wyllie (Reference Lee and Wyllie1998) commented ‘We see no prospect in the phase relationships that a parent natrocarbonatite liquid could follow a crystallization path toward residual calciocarbonatite’, as advocated by Le Bas (Reference Le Bas, Fitton and Upton1987), and more recently by a number of authors arguing this from the composition of fluid inclusions (Veksler et al. Reference Veksler, Nielsen and Sokolov1998a; Guzmics et al. Reference Guzmics, Mitchell, Szabó, Berkesi, Milke and Abart2011).
4. Experimental studies of liquid immiscibility using natural nephelinite
The Freestone and Hamilton investigations were followed by those of Baker and Wyllie (Reference Baker and Wyllie1990), Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991), Kjarsgaard et al. (Reference Kjarsgaard, Hamilton, Peterson, Bell and Keller1995) and Kjarsgaard (Reference Kjarsgaard1998), which are the most repeatedly cited studies in support of liquid immiscibility having occurred in natural nephelinite magmas. These studies sought to demonstrate that an ‘evolved nephelinite liquid’ will produce two contrasted silicate and carbonate liquids by liquid immiscibility.
Much of the reasoning for the experimental design is based on the presence of calcite-rich ‘globules’ described from the Suswa volcano in Kenya (Macdonald et al. Reference Macdonald, Kjarsgaard, Skilling, Davies, Hamilton and Black1993) and from the Shombole volcano in Kenya (Peterson, Reference Peterson1989; Kjarsgaard & Peterson, Reference Kjarsgaard and Peterson1991). Their similarity to the round calcites of the earlier experimental studies led to their being interpreted by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) as ‘quenched calcite melts’ and so indicative of liquid immiscibility. However, the globules in the Shombole nephelinites are not all monomineralic as they contain from 0 to 100% zeolites together with a number of other minerals. Peterson (Reference Peterson1989, p. 463) noted ‘The carbonate globules might be interpreted as immiscible carbonatite segregations or as secondary infilling of vesicles’. This caveat appears to have since been forgotten by proponents of liquid immiscibility. Textural features, such as the modally-zoned globules illustrated by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991), are more suggestive of a vesicular origin rather than an altered former silica-bearing calcitic carbonatite liquid. Clearly, these assemblages have little in common with the calcite spheres formed in the Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988, Reference Kjarsgaard, Hamilton and Bell1989) experiments and their proposed equivalence to these is unwarranted. Regardless, Peterson (Reference Peterson1989) rejected a vesicular origin and interpreted the textural and mineralogical features of the globules as quenched calcite melts. Consequently, Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) assumed the Shombole globules were formed by liquid immiscibility from a nephelinite magma.
Initial experiments on Shombole nephelinites by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) at temperatures of 975–925o C and 0.2 and 0.5 GPa are claimed to have reproduced the natural assemblages and that the carbonate-rich globules are quenched immiscible liquids. Furthermore, as the composition of minerals in the globules and the silicate groundmass are nearly identical, the samples were quenched when two liquids were in near equilibrium. These experimental data are further interpreted to suggest that the experimentally produced carbonates are equivalent to calcite carbonatites associated with nephelinitic magmatism. Assessment of the claims of Reference Kjarsgaard and Peterson Kjarsgaard and Peterson (1991) is difficult as no details of the compositions of the liquids involved are actually tabulated. For example, the experimentally determined liquidus line of descent of lava SH40 (Fig. 2), a titanite nephelinite, which contains 5.7 vol.% globules, on a T-X diagram at 0.5 GPa and 900–1025oC, cannot be readily evaluated as the interpretation relied on data for lava SH 49+10 wt.% calcite, which were not actually published until 1998 (see below). In particular, the oxide compositions of the silicate and carbonate liquids defining the abscissa (Lc and Ls in Fig. 2) are not specified, and the liquid compositions along the limbs of the solvus are not given. Note that the solvus for this composition is shown as symmetrical below 975oC and asymmetric above this temperature (Fig. 2). It is particularly important that the two-liquid T-X solvus for SH40 was not actually determined. In these experiments at 1025oC, one liquid exists and as temperature is decreased, clinopyroxene and nepheline become liquidus phases at 1015oC, crystallization of which leads to an increase in the carbonate component of the silicate liquid and entry at 960oC to the two-liquid field. What is not stated by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) is the actual composition of this proposed conjugate carbonate liquid, which cannot be the same as that determined for SH49+10 wt.% calcite (see below), and there is no information on how the immiscibility was recognized i.e. there are no BSE images of spherical carbonate globules. If present, these would, at that time, be considered to be calcite liquids, following Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989). However, our current interpretation would imply liquidus spherical calcite rather than a carbonated liquid. With further decrease in temperature, titanite and magnetite appear as sub-liquidus phases, plus the supposed immiscible carbonate, followed by calcite and sanidine at 915oC. There is no information on the textural relations of the calcite to the immiscible carbonate liquid and/or if this is formed interstitially to the silicate–oxide assemblage. It is on the basis of the experiments of Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991), coupled with those of Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989), that it was considered that calcite carbonatites are derived by liquid immiscibility from carbonated nephelinites and natrocarbonatites from peralkaline combeite nephelinites.
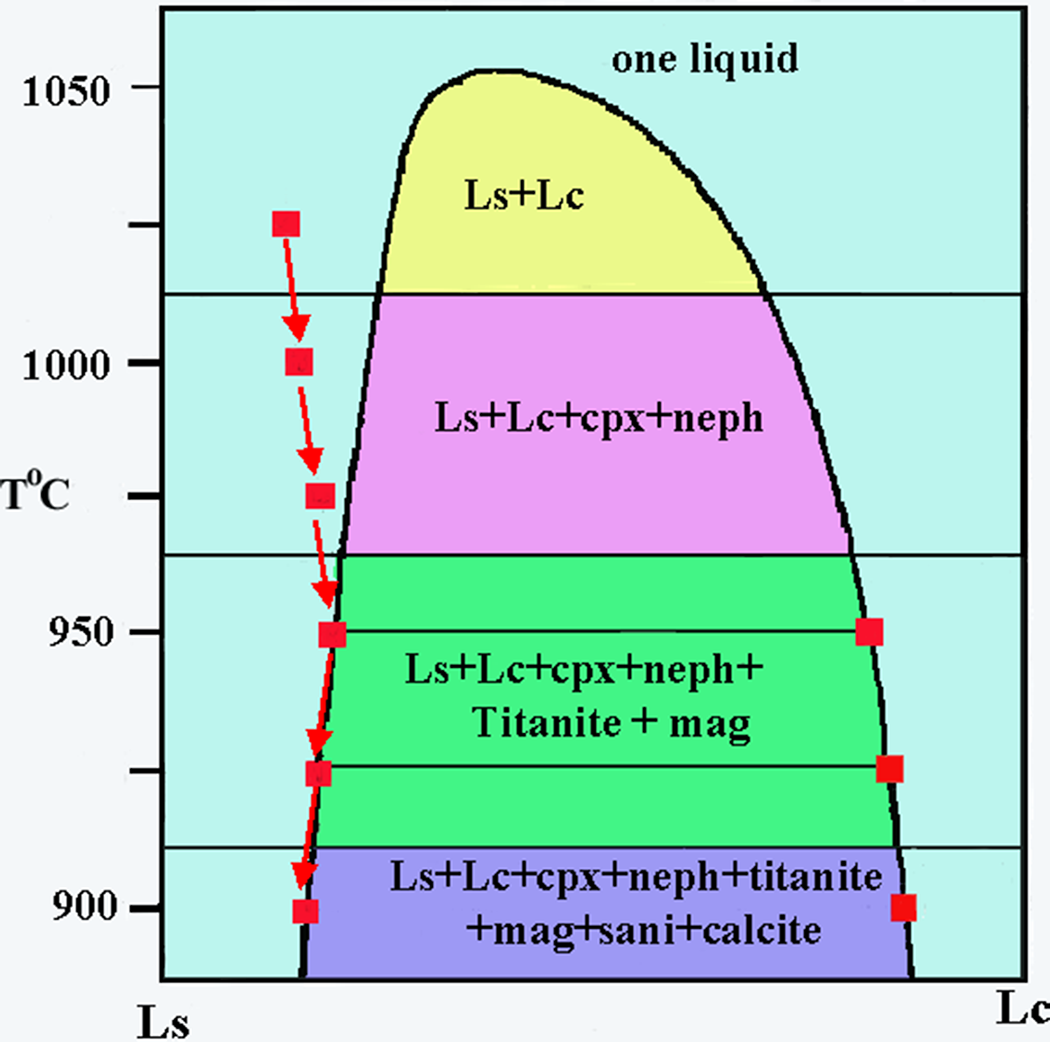
Figure 2. Experimentally determined schematic T-X solvus diagram for Shombole nephelinite SH40 at 500 MPa after Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991). The liquid line of descent from 1025–900oC is shown by the red arrows together with the compositions of co-existing silicate (Ls) and carbonate (Lc) immiscible liquids shown by the red squares. See the text for further details (cpx = clinopyroxene; neph = nepheline; sani = sanidine; mag = magnetite).
In seeking to prove this hypothesis, Kjarsgaard (Reference Kjarsgaard1998) undertook further experiments at 0.2 and 0.5 GPa and 1025 to 1040oC on Shombole perovskite nephelinite SH49, which contains 8.9 vol.% globules. Melting experiments were initially carried out on SH49 alone, although these data are not included in the 1998 paper and were never published. Apparently, globules of immiscible silicate and carbonate liquids were produced but were too small for accurate electron microprobe analysis. Hence, the experiments were repeated with the addition of 10 wt.% calcite to SH49 in order to produce larger globules (personal communication from Kjarsgaard). This increased the CaO content of SH49 from 12.6 to 16.6 wt.%; an unusually high value for nephelinites. It is apparent that not all of the added calcite dissolved in the melting experiments as there is a vapour phase in every experimental run. The listing of ‘F’ instead of ‘V’ in Table 2 of Kjarsgaard (Reference Kjarsgaard1998) is a misprint (pers.comm.). The presence of vapour in the unmodified SH49 experiments (Kjarsgaard, pers.comm) is particularly significant in that it shows that at least some of the calcite globules present in the lava represent calcite acquired after crystallization of the enclosing nephelinite magma. If these had truly exsolved by immiscibility, they should dissolve completely upon remelting; the presence of a vapour phase shows that this did not occur. This suggests that at least some of the calcite seen in SH49 is actually vesicle filling. If the 12.6% CaO contained in SH49 exceeds the amount that can remain in solution in the magma, then how much more so will 16.6% CaO exceed this level? No attempt was made to ascertain the maximum CaO content beyond which the nephelinite magma expels the excess as calcite-rich immiscible but not conjugate liquid. Although the experiments produced calcite-filled globules, these did not have the texture of those present in the lavas and consisted of quenched dendritic calcite. We interpret this to be a direct result of adding calcite to the nephelinite, a process that simply guarantees that an immiscible carbonate liquid will be expelled; thus it is purely a function of experimental design. We agree that silicate and carbonate liquids are inherently immiscible, but these experiments do not prove that they are conjugate or that nephelinite magmas undergo liquid immiscibility on cooling to form calcite carbonatites.
Brooker and Kjarsgaard (Reference Brooker and Kjarsgaard2011) have presented experimental studies of the system SiO2-Na2O–Al2O3–CaO–CO2 at 0.1–2.5 GPa and 1225–1700oC, which demonstrated the presence of a two-liquid field for most of these conditions. Although we do not doubt the veracity of these experiments, we consider them of interest only with respect to high-pressure experimental petrology because the bulk compositions used have little relevance to actual magma compositions as they used mixtures of silicate glasses with high CaO or Na2O contents, sodium carbonate, calcium carbonate and alumina. The absence of MgO and the extremely high temperatures involved are particular impediments in extrapolation to natural magmas. Because of these unrealistic experimental conditions, we consider that extrapolations by Brooker and Kjarsgaard (Reference Brooker and Kjarsgaard2011) to actual carbonatite-forming magma genesis are unwarranted.
Weidendorfer and Asimov (Reference Weidendorfer and Asimov2022) considered previously reported melting experiments conducted on modified nephelinite and synthetic systems to be invalid as the artificial bulk compositions used led to silicate liquids that were unrealistically enriched in alkalies, which failed to match the compositions of co-existing carbonatites and silicate rocks. Hence, they used starting materials, which are claimed to be ‘unmodified subvolcanic samples from Brava Island (Cabo Verde)’. They reported liquid immiscibility for a nephelinite occurring between 1100 and 950oC at 1.0 GPa. However, while they consider the starting composition (B52) to be unmodified, it is notable that it contains 8 wt.% CO2, surely implying that the nephelinite originally contained considerable amounts of calcite. It may not have been modified by Weidendorfer and Asimov, but it certainly has already been modified by nature. This was not a pristine fresh nephelinite that could reasonably be construed as representing an unmodified magma.
Furthermore, none of the samples investigated actually appear to represent liquids, and are not, in terms of their petrography, actually extrusive ‘nephelinites’, but their plutonic equivalents (ijolite series) whose compositions are determined by crystal accumulations. Figure 3 of Weidendorfer et al. (Reference Weidendorfer, Schmidt and Mattsson2016) clearly demonstrates this observation as fig. 3c, a supposed ‘nephelinite’, is petrographically a typical ijolite, and fig. 3d illustrates a calcite ijolite. It is notable that Weidendorfer et al. (Reference Weidendorfer, Schmidt and Mattsson2016, p. 43) recognize that many Brava rocks are cumulates and state ‘neither the true liquid composition nor a representative modal proportion of the fractionating minerals assemblage is represented by the intrusive (our italics) rock sample. Our liquid line of descent can thus only approximate the rock evolution’. We suggest that plotting the Brava bulk rock compositions on the IUGS-TAS diagram designed for volcanic rock classifications does not make them ‘liquid compositions’. Thus, contrary to Weidendorfer et al. (Reference Weidendorfer, Schmidt and Mattsson2016), there is actually no liquid line of descent but merely a curve of compositions representing modal variations in plutonic rocks. Importantly, there is no petrographic evidence of liquid immiscibility in any of the rocks investigated and calcite is clearly a late-stage mineral: a common feature observed in many plutonic ijolites. Thus, one is forced to conclude that the Weidendorfer and Asimov (Reference Weidendorfer and Asimov2022) experimental results are no more valid than those of Kjarsgaard (Reference Kjarsgaard1998) and Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) on which we have already cast considerable doubt.

Figure 3. Round liquidus calcite (cc) and fluorite (cf) in a matrix of quench calcite, fluorite and Ca2Nb2O7 formed in the quaternary system CaF2–CaCO3–Ca(OH)2–NaNbO3 at 800oC and 0.1 GPa (Mitchell and Kjarsgaard, unpublished; Mitchell Reference Mitchell2005b), which cannot be interpreted as former liquids. Similar round calcite was formed in experiments by Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989, 1998) and incorrectly interpreted as former liquids. Following that approach, the round fluorite would also be considered as an immiscible liquid and the quench matrix a third liquid.
Recent experiments by Lustrino et al. (Reference Lustrino, Luciani, Stagno, Narzisi, Masota and Scarlato2022) have reported the assimilation of up to 50 wt.% CaCO3 by melilititic and basanitic liquids at 1100o−1300oC and 0.2 GPa without liquid immiscibility occurring. A distinction must be drawn between ‘assimilation’ and ‘dissolution’ of CaCO3 since a vapour phase is present in all the experiments. The experiments show that a certain amount of the added CaCO3 (undetermined) actually dissolved, but the undissolved balance dissociated with the CaO component dissolving in the silicate liquid and the CO2 being released. In short, the solubility of CaO exceeds that of calcite. An important aspect of this work is that no immiscibility occurred. These experiments cast further doubt on the applicability of both Kjarsgaard (Reference Kjarsgaard1998) and Weidendorfer and Asimov (Reference Weidendorfer and Asimov2022) to carbonatite genesis and further undermine the establishment of liquid immiscibility in the evolution of nephelinitic magma.
We may conclude that no experimental study has yet demonstrated that nephelinite liquid can evolve to the stage where conjugate immiscible silicate and carbonatite liquids separate. However, Kjarsgaard (Reference Kjarsgaard1998) is widely cited as evidence that silicate and carbonatite liquid can separate immiscibly from nephelinite magma as conjugate pairs and has become the foundation of such claims. Hence, we have to question whether that ‘foundation’ has foundered. Even though we are also experimentalists, we are a little diffident to appear to cast aspersions on the extrapolation from noble metal tubes to outcrop, but we are inclined to think that some of the extrapolations have been a trifle incautious at times.
Ray et al. (Reference Ray, Ramesh, Pande, Trivedi, Shukla and Patel2000) have noted: ‘Liquid immiscibility may be the most important mechanism for the evolution of carbonatites and alkaline rocks but laboratory experiments to test this hypothesis, although demonstrating that the carbonatite and silicate “magmas” are immiscible, do not necessarily prove that they are of a common parentage’. Further, it is not always clear whether liquid immiscibility is considered to have developed as a supra-liquidus or sub-liquidus process. Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989) found both in their experiments. At 0.2 GPa, immiscibility occurred above the liquidus, but at 0.5 GPa, it occurred below the liquidus. Ray (Reference Ray1998) saw it as a sub-liquidus process and stated ‘Immiscibility initiates when the carbonate concentration of the parent (carbonated silicate) magma increases with the fractional crystallization of silicates’. Whereas most authors appear to consider that the two immiscible liquids completely separate and continue to their solidi, completely independent of each other. Ray (Reference Ray1998) saw it as a continuous process, writing ‘Irrespective of the initial carbonate content of the parent magma, which determines the timing of onset of liquid immiscibility, the fractional crystallization of silicates and the exsolution of carbonate liquid take place together and possibly might continue until the carbonate liquid is completely removed from the parent magma’. However, he further complicated the process by introducing the assimilation of basement gneiss; i.e. three simultaneous processes, as a third event. He visualized liquid immiscibility as an ongoing process that continues to develop during the protracted crystallization process. In general, it appears that sub-liquidus development of liquid immiscibility is the favoured scheme.
In summary, in terms of experiments, the only cases claiming to show the development of liquid immiscibility are those in which the starting material has been doctored by the addition of substantial amounts of calcite or sodium carbonate; either in the laboratory or by nature. In actuality, nephelinite magmas commonly appear to fractionate to produce phonolite or phonolitic nephelinite in volcanic systems or ijolite and/or malignite in plutonic settings (see below). In many instances, these magmas appear to have simply crystallized as nephelinite rather than evolving to the stage of separating into two contrasted conjugate liquids. Comments on the actual geological evidence invoked by liquid immiscibility advocates are considered below in Sections 9 and 10.
5. Round calcite crystals
Calcite crystals with a round morphology are common in experiments (Fig. 3). Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988, Reference Kjarsgaard, Hamilton and Bell1989), and earlier in oral communications, described these as ‘globules’ and considered that they crystallized from droplets of calcite liquid, hence demonstrating liquid immiscibility. For many years they were widely and uncritically accepted as such. Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989) illustrated calcite-rich globules from the Shombole nephelinite and, indeed, rounded calcite became the Holy Grail of those seeking to adduce liquid immiscibility in carbonatites. There was, however, no reason to have ever misinterpreted the texture. As long ago as the 1950s, it had been observed in experiments at the Pennsylvania State University and considered to be liquidus calcite, albeit of unusual habit (Gittins, Reference Gittins1973). A claim to have retracted the misinterpretation of the texture by Macdonald et al. (Reference Macdonald, Kjarsgaard, Skilling, Davies, Hamilton and Black1993) is hardly supported by a small change in their fig. 5b with no accompanying statement in the text. The actual correction was made by Bruce Kjarsgaard as a personal communication in Lee et al. (Reference Lee, Wyllie and Rossman1994, p. 1136). The delay is unfortunate for it has contributed to the very widespread acceptance of rounded calcite as irrefutable evidence of liquid immiscibility being the dominant process of carbonatite-forming magma genesis. Importantly, Otto and Wyllie (Reference Otto and Wyllie1993) reported that in their experiments round calcite was always a primary liquidus crystalline phase, whereas prismatic calcite was always a quenched phase. Lee et al. (Reference Lee, Wyllie and Rossman1994) and Lee and Wyllie, (Reference Lee and Wyllie1994, Reference Lee and Wyllie1997, Reference Lee and Wyllie1998), responding to the problem, stated ‘We do not see how immiscible liquids with compositions near pure CaCO3 could be generated under any conditions’. There was even an illustration of the texture labelled as a ‘primary liquidus phase’ in Gittins (Reference Gittins1973; fig. 1) some twenty years before the Kjarsgaard’s revision of the origin of the texture. The misinterpretation also failed to note the well-established evidence from experimental studies that calcite cannot exist as a liquid at petrologically reasonable temperatures and pressures without the presence of fluxes such as alkalis, H2O or fluorine. Furthermore, pure calcite liquid could not, without the additional presence of elements such as Na, K, Si, Al, Fe and P, among others, produce the typical mineralogy of carbonatite rocks. Yet the concept dies hard; even today, Pirajno and Yu (Reference Pirajno and Yu2022, fig. 4) illustrate ‘immiscible calcite spherules in silicate liquid from the Goudini carbonatite’ in complete disregard for all the evidence against the concept, and they and Hurai et al. (Reference Hurai, Huraiova, Habler, Horschinegg, Milosvky, Milovska, Hain and Abart2022) interpret globular carbonates as quenched carbonatitic liquids without any reference to Lee et al. (Reference Lee, Wyllie and Rossman1994).
Interestingly, there are still no explanations of why minerals which normally crystallize with well-defined crystal faces should form round crystals in experiments but never in natural systems. The phenomenon has been recognized for calcite (Kjarsgaard & Hamilton, Reference Kjarsgaard, Hamilton and Bell1989; Otto & Wyllie, Reference Otto and Wyllie1993; Lee et al. Reference Lee, Wyllie and Rossman1994; Mitchell, Reference Mitchell, Linnen and Samson2005a; Chebotarev et al. Reference Chebotarev, Veksker, Wohlgemuth-Ueberwasser, Doroshkevich and Koch-Müller2019), fluorite (Mitchell and Kjarsgaard, unpublished here as Fig. 3; Chebotarev et al. Reference Chebotarev, Veksker, Wohlgemuth-Ueberwasser, Doroshkevich and Koch-Müller2019) and nyerereite (Nikolenko et al. Reference Nikolenko, Stepano, Roddatis and Veksler2022).
6. Trace elements and partition coefficients
Many proponents of a liquid immiscibility origin for carbonatite-forming magma have suggested that there is a similarity between experimentally determined partition coefficients and the bulk rock trace element contents of carbonatites and silicate rocks in carbonatite–alkaline-rock complexes. As these ‘partition coefficients’ have been determined on what are claimed to be conjugate pairs of liquids, it has been deemed that this similarity proves that the two contrasted magmas are formed by liquid immiscibility. Studies of partition coefficient determination start by assuming that the immiscible liquids in their experiments are conjugate. We have already argued above that although these liquids are immiscible in a geological context, they are not conjugate; i.e. they are not generated by the immiscible separation of a single-phase liquid.
Many of the studies involve compositions related to those of Oldoinyo Lengai nyerereite–gregoryite lavas and so are of extremely limited applicability to commoner types of carbonatite. Others try to approach more realistic calcic carbonatite compositions (Martin et al. Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012) but the same weakness in the initial assumptions remains. Are these really ‘partition’ coefficients or simply an expression of the geochemical affinities for which elements prefer which liquid composition, regardless of how these originate?
The experimental design of the studies may be questioned on two grounds. The most important is the basic assumption that conjugate liquid immiscibility occurs. But this is never proven in any of the experiments by taking the charges to temperatures above the two-liquid solvus and cooling to temperatures at which immiscibility actually occurs. Further, the temperatures involved are typically very high (>1100oC) and unlikely to be achieved in natural alkaline magmas.
The experiments can also be considered examples of the well-known chemical process of solvent extraction; in this case between a polymerized silicate liquid and an ionic carbonate liquid. Typically, the experiments do not follow normal solvent extraction methods, which are dynamic and involve several episodes of agitated rheological mixing of the two liquids involved followed by density-controlled final separation. This process effects the maximum distribution of elements between the two liquids. Simple static equilibration of two liquids cannot effect total extraction of trace elements by diffusional processes. Centrifuge experiments actually exacerbate this effect as the liquids are rapidly separated. The experimental problems might be resolved if experiments were undertaken using rocking autoclaves, but unfortunately such equipment capable of operating at high temperatures and pressures is not yet available.
Hamilton et al. (Reference Hamilton, Bedson, Esson and Bell1989) undertook experiments at 1150o and 1250o and 0.1–0.6 GPa using phonolite (4.91 K2O; 2.67 CaO; 10.63Na2O wt.%) or nephelinite (4.95 K2O; 10.89 CaO; 10.6 Na2O wt.%) mixed with K–Ca–Na carbonates (8.34 K2O ; 16.69 CaO; 33.38 Na2O wt.%) or synthetic calcic carbonatite (5.55 K2O; 33.32 CaO; 16.6 Na2O wt.%), respectively. The admixed carbonate composition richer in CaO corresponds to the carbonated liquid found by Freestone and Hamilton (Reference Freestone and Hamilton1980) to be in equilibrium with a silicate liquid of similar composition to that of a nephelinite from Oldoinyo Lengai. Hamilton et al. (Reference Hamilton, Bedson, Esson and Bell1989) concluded that enrichments of trace elements (Ba, REE, Hf, Ta, Zr) into carbonate liquids is favoured by high pressures, low temperatures and increased polymerization of the silicate conjugate melt. The formation of carbonatites enriched in incompatible elements is considered to be a multi-stage process in which liquid immiscibility occurs in a carbonated alkali-rich magma in the upper mantle, followed by crystallization of calcite from the carbonated liquid leading to further enrichments in trace elements. Although of relevance to Oldoinyo Lengai, these experiments have no relevance to the formation of calcite carbonatites and the partition coefficients should not be cited as such.
Veksler et al. (Reference Veksler, Petibon, Jenner, Dorfman and Dingwell1998b), using centrifugal experiments for phase separation, investigated bulk compositions in the synthetic system, SiO2–Al2O3–CaO–Na2O–K2O at 965o and 1015o at 0.085–0.092 GPa, that were extremely enriched in Na2O (∼22–33 wt.%) and K2O (∼ 13 wt.%), and poor in CaO (∼7–11 wt.%). The proportions of aluminosilicate to carbonate in the starting compositions were completely arbitrary e.g. (44 wt.% (SiO2+Al2O3) plus 56 wt.% (Na–Ca–K carbonates). In all cases, the quenched runs contained two liquids but no runs were at a high enough temperature to create a single-phase liquid. Crystalline liquidus phases were not found in any of the experiments, apart from trace amounts of combeite in the silicate liquid of only one of the low-temperature (965oC) experiments. Although two immiscible liquids were formed, their conjugate status was not established. Clearly, the distribution coefficients obtained for trace elements between these synthetic silicate and carbonate liquids are irrelevant to natural systems. These distribution coefficients, and in particular the observation that Ba is preferentially concentrated in carbonate liquids whereas Zr is concentrated in silicate liquids, have been used (e.g. Martin et al. Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012; de Moor, Reference de Moor, Fischer, King, Botcharnikov, Hervig, Hilton, Barry, Mangasini and Ramirez2013; Halama et al. Reference Halama, Vennemann, Sebel and Markl2005; Mourão et al. 2012) to suggest liquid immiscibility relationships between silicate rocks and diverse carbonatites regardless that the bulk compositions of the rocks investigated are far removed from those of the experimental liquids and do not actually represent liquid compositions. Curiously, Veksler et al. (Reference Veksler, Petibon, Jenner, Dorfman and Dingwell1998b) suggested that the determined strong preference of Zr and Nb for the silicate liquid does not support the origin of Nb- and Zr-rich carbonatites by liquid immiscibility and suggests their formation by fractional crystallization. Subsequently, Veksler et al. (Reference Veksler, Dorfman, Dulski, Kamenetsky, Danyushevsky, Jeffries and Dingwell2012) extended this study by the addition of halides and sulfate to the starting compositions to investigate melts analogous to Oldoinyo Lengai nyerereite–gregoryite lavas. Interestingly, this study concluded that comparison of the experimentally determined partition coefficients and the bulk rock trace elements content of the nyerereite–gregoryite lavas and nephelinite reveals significant discrepancies thus rendering a simple single immiscibility model for Oldoinyo Lengai questionable.
In contrast, to other studies, Martin et al. (Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012) conducted centrifuge experiments on a series of Si-and Na-poor (21–33 wt.% SiO2; 0.3–2 wt.% Na2O), CaO-rich (12–23 wt.% CaO) bulk compositions at 1180–1250oC and 1.7 GPa to simulate element partitioning between immiscible carbonatite and kamafugite-like melts. The premise of the work is that the kamafugites and carbonatites of the Intra-Apennine Magmatic Province both originate in the upper mantle from unspecified primitive mantle CO2-bearing magmas by liquid immiscibility, as claimed by Stoppa and Lupini (Reference Stoppa and Lupini1993) and Stoppa et al. (Reference Stoppa, Rosatelli, Wall and Jeffries2005). The bulk compositions used in the experiments are based upon a synthetic mixture of oxides and carbonates corresponding to 50 wt.% of primitive San Venanzo kamafugite and 50 wt.% Polino carbonatite. We consider that this premise, and thus the conclusions of Martin et al. (Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012), is based on circular reasoning as there is no a priori reason why kamafugites and the geographically associated carbonatites should be derived from some common parental magma and/or be related in the proportions used in the experiments.
Although the experiments did demonstrate for the bulk compositions used, that the silicate and carbonate liquids were immiscible, it was never proven that these originated as conjugate melts separating from a single melt. Again it would appear that the bulk compositions used, particularly the unjustifiably large amount of carbonate, pre-determines a result of immiscible carbonate melt. The experimental silicate melt compositions obtained were found, not surprisingly, to be similar to those of San Venanzo kamafugite lava and the carbonate liquids to the hypothetical former compositions of the now extensively-altered calcitic carbonatites at Polino and Cupaello. Although these experiments suggest that kamafugite and carbonatite melts are not miscible, they certainly do not prove formation from a common parent. If the latter had a composition similar to the bulk compositions used in the experiments, it would be of a most unusual composition, and/or derived from decidedly unusual source rocks. Martin et al. (Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012) added a wide variety of trace elements to their starting compositions and reached similar conclusions regarding distribution coefficients as Veksler et al. (Reference Veksler, Petibon, Jenner, Dorfman and Dingwell1998b; Reference Veksler, Dorfman, Dulski, Kamenetsky, Danyushevsky, Jeffries and Dingwell2012) in that alkaline earths and rare earth elements (REE) were weakly concentrated in the carbonate melt and that Zr, Ti and Nb are enriched in the silicate melts.
Subsequently, Martin et al. (Reference Martin, Schmidt, Mattsson and Guenther2013) used a Si-poor (17 wt.% SiO2) modified nephelinite composition, based on that used by Lee and Wyllie (Reference Lee and Wyllie1997) with the composition of liquidus olivine subtracted. Again 50:50% silicate–carbonate proportions were used in which the carbonate component was 25% Na2CO3 and 25% dolomite. This starting mixture has 23 wt.% CO2; an extraordinarily high content relative to that of any naturally occurring silicate magma. Given that silicate and carbonate liquids are by their physical properties immiscible, as in the experiments of Martin et al. (Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012), the excess carbonate is expelled as an immiscible, but not conjugate, liquid from the silicate liquid, which at 1 GPa and 1240o C not surprisingly approaches that of the compositions and proportions of the starting nephelinite and carbonates i.e. ∼53 and ∼46 vol.%. Again, we consider that melting the starting compositions at temperatures below where a single-phase liquid exists is not appropriate. Determination of the distribution of trace elements between the two immiscible liquids encountered in the experiments is not a measure of the distribution of elements between conjugate liquids exsolved from a single-phase liquid. Martin et al. (Reference Martin, Schmidt, Mattsson and Guenther2013) also stated that ‘Increased polarization of the silicate melt leads to a shift of trace element partitioning toward the carbonatite melt, as trace elements become increasingly incompatible with silicate melt polymerization’. This is a fundamental property of silicate and carbonate liquids and is equally true regardless of how the liquids are developed. It does not prove conjugate liquid immiscibility. Thus, Martin et al. (2012, Reference Martin, Schmidt, Mattsson and Guenther2013) experiments on both nephelinite–carbonate and further studies of kamafugite–carbonate mixtures are actually of no relevance to the development of liquid immiscibility in natural systems. The distribution coefficients determined represent equilibria between immiscible polymerized silicate and polar carbonate liquids and not those of conjugate liquid-liquid immiscibility.
Guzmics et al. (Reference Guzmics, Zajacz, Mitchell, Szabó and Wälle2015) have provided a set of trace element distribution coefficients derived from ‘crystalline melt inclusions’ occurring in nepheline from Kerimasi afrikandite. The inclusions were heated (up to 1100oC) and quenched at atmospheric pressure. Guzmics et al. (Reference Guzmics, Zajacz, Mitchell, Szabó and Wälle2015) did not demonstrate that the inclusions were initially heated to temperatures above any solvus consolute temperature. Considerable differences exist with respect to the partition data of Veksler et al. (Reference Veksler, Dorfman, Dulski, Kamenetsky, Danyushevsky, Jeffries and Dingwell2012) and Martin et al. (Reference Martin, Schmid, Mattsson, Ulmer, Hametner and Guenther2012, Reference Martin, Schmidt, Mattsson and Guenther2013). These differences, in particular with regard to Pb, Zr, Nb and Ta partition, are considered to reflect differences in the compositions of this natural system containing more Ca and P relative to the more sodic experimental systems. Whereas, again this investigation does not prove conjugate silicate–carbonate liquid immiscibility, it does indicate that values of distribution coefficients, apart from temperature, are very susceptible to the liquid bulk compositions. Hence, we advise that those who wish to use partition coefficients in genetic model computations should be wary of potentially significant errors in their calculations.
More recently, Nabyl et al. (Reference Nabyl, Massuyeau, Gaillard, Tuduri, Marziano, Roerie, Le Trong, DiCarlo, Melleton and Bailly2020) sought to determine the partition coefficients of some trace elements in co-existing purportedly conjugate silicate and carbonate immiscible liquids. These experiments were conducted using the same nephelinite to carbonatite proportions as did Kjarsgaard (Reference Kjarsgaard1998), namely synthetic nephelinite 90 wt.% and calcite 10 wt.%, with 0.1 wt.% Ba, Sr, Nb and REE added. That is to say, the experiments were on ‘nephelinite’ that was artificially enriched in calcite, a process which, as we have already pointed out, guarantees the production of two liquids that are immiscible but not necessarily conjugate. The experiments were run at 725 to 975oC and 0.2 to 1.5 GPa. The mixtures were first heated to 1000oC for the lower temperature runs and 1100oC for the higher temperature runs, allegedly to ensure that they were completely liquid before the temperature was lowered to the temperatures of the experimental runs. However, this assumption, and the Kjarsgaard (Reference Kjarsgaard1998) experiments, casts considerable doubt over whether these temperatures were high enough to achieve complete melting.
Regardless, the runs at various temperatures were quenched and the two former liquids were analyzed to determine partition coefficients. The authors infer that the two immiscible liquids continued to exchange components as they cooled and then proceeded to equate the changing silicate liquid compositions from nephelinite to phonolite, with those of the corresponding rocks in naturally occurring carbonatite complexes. They conclude that these experiments model the behaviour of evolving magmas. But this deduction falsely equates the behaviour of liquids in a small noble metal tube with those in an active igneous complex. In the experiment, the liquids are in intimate contact throughout the cooling period during which they might be able to exchange components, but this cannot be the situation in any putative magma chamber where in order to be intruded as contrasted rock types, they must first have separated into completely discrete bodies of magma. They are then no longer in intimate contact and are prevented from exchanging components. It is indeed dubious whether, even in the confines of the experiment, an inter-liquid exchange actually occurs. It seems more probable that the two liquids continue their evolution independently by fractional crystallization. The gradual increase in REE content of the carbonatite liquid is most probably due to the progressive crystallization of calcite, thus causing the increase of the REE content of the residual liquid to increase, rather than from continuing transfer from the evolving silicate liquid. Clearly, the extrapolation by Nabyl et al. (Reference Nabyl, Massuyeau, Gaillard, Tuduri, Marziano, Roerie, Le Trong, DiCarlo, Melleton and Bailly2020) of progressively evolving liquid compositions in experiments to those of the constituent rock types of a carbonatite complex, even if it did occur, is inappropriate. The experimental design is based on an unwarranted assumption and reaches conclusions that are both invalid and seriously misleading for the understanding of carbonatite petrogenesis.
The experiments of Nabyl et al. (Reference Nabyl, Massuyeau, Gaillard, Tuduri, Marziano, Roerie, Le Trong, DiCarlo, Melleton and Bailly2020) also have some inconsistencies as they used the same starting compositions as Kjarsgaard (Reference Kjarsgaard1998), and one might therefore expect their experimental results to be comparable. However, their carbonate liquid at 925oC has 5–13 wt.% more SiO2 and half the Na2O content as that of Kjarsgaard (Reference Kjarsgaard1998). A further ambiguity is related to fig. 1 of Nabyl et al. (Reference Nabyl, Massuyeau, Gaillard, Tuduri, Marziano, Roerie, Le Trong, DiCarlo, Melleton and Bailly2020), which shows crystalline calcite in the silicate liquid fraction. The origin of this calcite is not explained but it is certain that it will contain some of the trace elements, which Nabyl et al. (Reference Nabyl, Massuyeau, Gaillard, Tuduri, Marziano, Roerie, Le Trong, DiCarlo, Melleton and Bailly2020) consider to be partitioned into the carbonate fraction; hence the distribution coefficients determined might be in error.
The underlying assumption of all the experimental determinations of partition coefficients described above is that the immiscible silicate and carbonate liquids are conjugate. We suggest that measuring the trace element bulk compositions of co-existing carbonatite and silicate rocks and then arguing that the results are consistent with the partitioning experiments, thus proving liquid immiscibility, constitutes circular reasoning. More problematic is that most carbonatites do not have bulk compositions representative of liquids and many are demonstrably cumulates. Of course, the carbonatite-forming magma must have acquired its unique trace element composition at some stage in its evolutionary history, but whether it happens during partial melting of the mantle or at some later stage is not, and cannot be, determined by measuring the trace element distribution between hypothetical liquids whose existence in nature is a contrived assumption. Again there is confusion between conjugate liquid immiscibility and liquids that are immiscible but not necessarily conjugate. Our conclusion is that there are no determinations of trace element partitioning, which are relevant to the genesis of calcite or dolomite carbonatite and even those concerned with nyerereite–gregoryite lavas are unrealistic.
7. Hypotheses based on isotopic similarity
Immiscibility models require that both carbonate and silicate liquids be in chemical and isotopic equilibrium. Consequently, many investigations state that if the Sr and Nd isotopic compositions of the carbonatite and accompanying silicate rocks are similar, both rock types must have a common source, and liquid immiscibility must be the explanation for this similarity. This is a gravely overstated and invalid assumption. Certainly, there is no a priori geological reason why isotopic similarity is proof of carbonatites and silicate rocks being derived from a common parental liquid. Unfortunately, many investigations of the isotopic compositions of carbonatites (e.g. Rukhlov et al. (Reference Rukhlov, Bell, Amelin, Simandl and Neetz2015) have not included determination of those of the associated silicate rocks and have concentrated solely on explanations of the mantle origins of the isotopic variations.
In some carbonatite complexes, the isotopic data are consistent e.g. Prairie Lake (Wu et al. Reference Wu, Mitchell, Li, Zhang and Yang2017) or Aillik Bay (Tappe et al. Reference Tappe, Foley, Jenner, Heaman, Kjarsgaard, Romer, Stracke, Joyce and Hoefs2006), whereas others are inconsistent e.g. Spitskop (Harmer Reference Harmer1999), with their having a common origin. Bell (Reference Bell1998) commented that the silicate rocks commonly show much greater variation in isotopic composition than their associated carbonatites, with more examples where the isotopic ratios are distinctly different. Examples cited by Harmer and Gittins (Reference Harmer and Gittins1998) include Kerimasi (Tanzania) where the carbonatites are isotopically similar to melilitite lava and an ijolite intrusive, but distinct from the nephelinite and phonolitic nephelinite lavas, which have Sr and Nd isotopic ratios significantly higher and lower than the carbonatites. At Shombole (Kenya), the carbonatites have similar ranges of Sr and Nd isotopic ratios to those of the nephelinites, whereas the phonolites are distinctly different (Bell and Peterson Reference Bell and Peterson1991). Thus, the carbonatites could be derived from the nephelinites, as argued by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991), but not from the phonolites. At Spitskop (Harmer, Reference Harmer1999), the dolomitic carbonatites have compositions similar to the calcite carbonatites. Both are different from those of the associated ijolites and thus could not have been derived from a single-parent magma by either fractional crystallization or liquid immiscibility. Similarly, the dolomitic Dorowa and Shawa carbonatite complexes in Zimbabwe (Harmer et al. Reference Harmer, Lee and Eglington1998) show that the carbonatites and accompanying nephelinites represent magmas that were derived from different portions of the subcratonic mantle of southern Africa and that the Shawa carbonatites must have been derived from a greater depth than the silicate rocks. Harmer and Gittins (Reference Harmer and Gittins1998) wrote ‘If immiscibility was involved in the generation of these carbonatites, it is constrained to have occurred at an early stage in the evolution of the parental magmas; from less, rather than from more evolved, silicate magmas (i.e melilitite or olivine nephelinite) rather than phonolite’. The inescapable conclusion is that while some carbonatite may be derived from silicate magma, it could only be from some, not all, of the silicate magma involved in the formation of the complex. At Aillik Bay, Tappe et al. (Reference Tappe, Foley, Jenner, Heaman, Kjarsgaard, Romer, Stracke, Joyce and Hoefs2006) have shown that dolomite carbonates and ultramafic lamprophyres have similar Sr and Nd isotopic compositions and have claimed that these rock types are related by liquid immiscibility. However, these isotopic data actually have no bearing on the process of immiscibility as actually this was proposed using other evidence. This included interpretation of ambiguous textures, appeal to the Hamilton projection and use of the Veksler et al. (Reference Veksler, Petibon, Jenner, Dorfman and Dingwell1998b) distribution coefficients.
A recently published isotopic study of the Finca La Nava complex in the Calatrava volcanic province of Spain (Rosatelli et al. Reference Rosatelli, Humphreys-Willians, Wall, Castorini, Perna and Stoppa2022) displays the importance of carefully comparing the isotope compositions of the contrasted rock types in a complex. The authors found that the carbonatites have Sr and Nd isotopic compositions which differ from those of the melilitite nephelinites and conclude that ‘the melilite nephelinites and carbonatites are co-eruptive but not co-magmatic’. Clearly, liquid immiscibility is ruled out.
The differences in isotopic composition between silicate rocks and carbonatites in a given complex are commonly explained by diverse mantle and/or crustal contamination processes. It is typically assumed that silicate rocks are more susceptible to contamination because of their lower Sr and REE contents relative to associated carbonatites. Although immiscible carbonate liquids could be susceptible to similar degrees of contamination subsequent to liquid immiscibility, and that this might not be recognizable, it is not reasonable to expect these diverse magmas to interact with potential contaminants in the same manner. Further, it has been demonstrated that carbonatites in some cases exhibit significant Sr and/or Nd isotopic variation, such as at Jacupiranga (Roden et al. Reference Roden, Murthy and Gaspar1985) and Spitskop (Harmer Reference Harmer1999). These data suggest that even Sr and REE-rich carbonatites could be either contaminated or reflect isotopic variation in their source rocks. Post-immiscibility contamination of conjugate silicate rocks can be ruled out as these should exhibit even greater isotopic heterogeneity. We conclude that even in cases where the radiogenic isotopic ratios of the silicate and carbonate rocks are similar, this does not constitute evidence of liquid immiscibility.
8. Melt inclusions
Much attention has been given to the hypothesis that ‘melt inclusions’ found in the major mineral phases of plutonic ijolites, pyroxenites and calcite carbonatites are representative of contemporaneous cogenetic magma (Veksler et al. Reference Veksler, Nielsen and Sokolov1998a; Guzmics et al. Reference Guzmics, Zajacz, Mitchell, Szabó and Wälle2015; Chayka et al. Reference Chayka, Kamenetsky, Vladykin, Kontonikas-Charos, Prokopyev, Stepanov and Krasheninnikov2021; Prokopyev et al. Reference Prokopyev, Doroshkevich, Zhumadilova, Starikova, Nugumanova and Vladykin2021). However, the majority of these inclusions are not quenched glasses and consist of diverse assemblages of crystalline carbonates, phosphates, magnetite and silicates (commonly monticellite) with non-quenchable Na-Ca carbonates similar to nyerereite or shortite. That these assemblages actually crystallized from a melt is not proven; hence the term is actually a misnomer. Certainly, inclusions consisting solely of apatite and Na–Ca carbonatite, as illustrated by figs. 5c and 5d of Guzmics et al. (Reference Guzmics, Mitchell, Szabó, Berkesi, Milke and Abart2011) cannot represent any realistic parental melt. Interestingly, similar assemblages of minerals that occur as inclusions in calcite in volcanic carbonatites at Kerimasi cannot represent ‘melt inclusions’ (Mitchell & Dawson, Reference Mitchell and Dawson2021). For an as yet unexplained reason, unambiguous melt inclusions containing quenched silicate glass co-existing with quenched Na–Ca carbonate are found only in nepheline from Oldoinyo Lengai combeite wollastonite nephelinite (Mitchell, Reference Mitchell2009) and Kerimasi afrikandite (Guzmics et al. Reference Guzmics, Zajacz, Mitchell, Szabó and Wälle2015). In many instances, the ‘crystallized melt inclusions’ coexist with bona fide low-temperature fluid inclusions.
Attempts have been made to reconstitute the supposed parental melts of the inclusions by heating to temperatures up to 1100oC at atmospheric pressure, followed by rapid quenching (Guzmics et al. Reference Guzmics, Mitchell, Szabó, Berkesi, Milke and Abart2011, Reference Guzmics, Mitchell, Szabó, Berkesi, Milke and Ratter2012, Reference Guzmics, Zajacz, Mitchell, Szabó and Wälle2015; Chayka et al. Reference Chayka, Kamenetsky, Vladykin, Kontonikas-Charos, Prokopyev, Stepanov and Krasheninnikov2021). In many of these experiments, a quenched silicate melt, with or without a carbonate, is produced. While we do not question the veracity of the experiments, we consider re-interpretation of the results is desirable, bearing in mind the character of the mineral assemblages present prior to heating, which might, or might not, represent the bulk composition of a parental melt due to heterogeneous trapping, local disequilibrium and/or reaction with host minerals (Veksler et al. Reference Veksler, Nielsen and Sokolov1998a). Although the bulk composition of individual inclusions is not known (or even estimated), heating of heterogeneous silicate, phosphate and Na–Ca–carbonate assemblages is analogous to the Kjarsgaard and Hamilton (Reference Kjarsgaard, Hamilton and Bell1989, 1998) experiments on synthetic silicate rock compositions with added Na2CO3. Not surprisingly, similar results are obtained on quenching the melted inclusions, as the silicate, carbonate and even sulphide liquids produced are inherently immiscible, but not necessarily conjugate. Although two, and three phase, immiscibility textures are found in the experimental run products (Guzmics et al. Reference Guzmics, Mitchell, Szabó, Berkesi, Milke and Ratter2012), depicting these melt compositions in the Hamilton projection does not prove liquid immiscibility has occurred, as this was never actually established by heating the inclusion to temperatures above the liquid solvus consolute temperatures, which extrapolate from the temperatures of the experiments, must be extraordinarily high.
We consider that ‘crystalline melt inclusions’ do not represent crystallized parental melt, although they do record the evolution of their host magmas by the changes in the compositions of the minerals present in them, as shown by Veksler et al. 1989 for the Kovdor and Gardiner complexes. Consequently, we regard the inclusions as being formed by the entrapment of pre-existing and/or current liquidus phases plus carbonate-rich residual fluid. As such, the ‘melt inclusions’ can be considered as an early part of a process of inclusion formation in conditions ranging from high temperatures to consanguineous bona fide low-temperature fluid-dominated inclusions (Chayka et al. Reference Chayka, Kamenetsky, Vladykin, Kontonikas-Charos, Prokopyev, Stepanov and Krasheninnikov2021; Prokopyev et al. Reference Prokopyev, Doroshkevich, Zhumadilova, Starikova, Nugumanova and Vladykin2021); the latter being termed the ‘brine melt stage’ by Anenburg et al. (Reference Anenburg, Broom-Fendley and Chen2021). In summary, ‘crystallized melt inclusions’ do not provide any evidence for liquid immiscibility in the genesis of carbonatites, although they are useful in understanding magmatic processes. We are in agreement with Kamenetsky et al. (Reference Kamenetsky, Doroshovich, Elliot and Zaitsev2021, p. 312) ‘that the use of multiphase inclusions has so far failed to adequately address (sic) the original compositions of carbonatite magmas’.
9. Critique of the geological evidence offered by advocates of liquid immiscibility
The principal concerns in determining the role of immiscibility in nature are: (1) Do natural magmas evolve to compositions at which immiscibility occurs; (2) At what stage of magmatic evolution does immiscibility occur i.e. high pressure and temperatures in the asthenospheric or lithospheric mantle, e.g. Wyllie (Reference Wyllie and Bell1989) or relatively low pressures and temperatures in a crustal environment, e.g. Le Bas (Reference Le Bas, Fitton and Upton1987); (3) What is the geological evidence for the process? We have discussed above some aspects of the first two of these concerns in our review of experimental evidence for immiscibility and will confine this discussion mainly to geological aspects of the process.
Proponents of liquid immiscibility in carbonatite genesis never actually discuss the mechanisms and rheology of the process. It is as if a magic wand is waved and perfect separation generates discrete magma bodies. Indeed Jones et al. (Reference Jones, Walker, Pickett, Murrell and Beattie1995) have done exactly that in advocating ‘instantaneous liquid immiscibility’ for Oldoinyo Lengai natrocarbonatite. The reality is that the initial stages of separation must surely lead to a ‘froth’ of carbonate liquid bubbles in a silicate liquid (or vice versa). To become two distinctly different liquids, this ‘froth’ must have both the time and space to separate completely into discrete bodies of magma. Such a process will require the existence of a suitable ‘magma chamber’, in which this separation can take place.
It is never explained why immiscibility always proceeds to the complete separation of the two liquids. Is it too much to expect that in all of the many hundreds of known carbonatite complexes, not one has been found to contain large volumes of ‘hybrid’ rocks resulting from extrusion or intrusion of a partially-separated magma? An additional major flaw in the liquid immiscibility claim is that when alkaline silicate rocks are accompanied by carbonatites, the latter are almost invariably the latest intrusions. There is a general acceptance that carbonatite-forming magmas should have viscosities orders of magnitude below those of silicate magmas, as well as having lower specific gravities than mantle and crustal rocks. Thus, they might be expected to ascend more readily than any silicate magma. However, if they are to comply with the physical constraints, the silicate-forming magmas must remain at depth until after the carbonatite-forming silicate has erupted. This is contrary to observations and expectations.
Furthermore, it is apparent that the majority of plutonic alkaline rock-carbonatite complexes appear to have been created by multiple intrusions of dikes and sills rather than crystallizing as a distinct single body of rock that might be expected of a ‘magma chamber’. Indeed, there is now a disinclination to accept the generality of the magma chamber hypothesis in favour of episodic intrusion of small batches of magma as sills (Cashman et al. Reference Cashman, Sparks and Blundy2017). An example of this new approach to emplacement processes is the Rum complex (Scotland), which consists of a layered suite of ultrabasic rocks with features similar to those seen in clastic sedimentary rocks, including layering and graded bedding. Previous research (Wager & Brown, Reference Wager and Brown1968) concluded that the layered suite was the result of events occurring in a magma chamber. However, recent research (Emeleus and Troll Reference Emeleus and Troll2014; Hepworth et al. Reference Hepworth, Kaufmann, Hecht, Geertisser and O’Driscoll2020) has questioned this hypothesis, and it is now proposed that the layered suite originated from repeated small sill-like intrusions into previously intruded material and that a magma chamber did not exist. With respect to carbonatite complexes, Savard and Mitchell (Reference Savard and Mitchell2021) have concluded that there is no geological evidence for the presence of a large-scale magma chamber at the Prairie Lake complex, and that ijolites and other rocks are formed by the crystallization of repeated injections of small batches of ijolite-forming magma into consolidated or partially-crystallized previously emplaced batches of magma of similar, but not identical, composition. Intrusion of magma into previously consolidated rocks can result in subsolidus recrystallization of the latter to form annealed ijolites, whereas intrusion into partially-consolidated material can result in filter pressing and mobilization of residual fractions such as carbonatite-forming liquids. In the volcanic environment, repeated injection of sill-like intrusions and filter-pressing of nyerereite–gregoryite lava has been suggested for Oldoinyo Lengai by Lundstrom et al. (Reference Lundstrom, Hervig, Fischer, Sivaguru, Yin, Zhou, Lin and Gros-Diniz2022).
Advocates of liquid immiscibility usually point to the ijolite–carbonatite association found in many plutonic complexes as evidence of bimodal magmatism, and hence liquid immiscibility (Le Bas, Reference Le Bas, Fitton and Upton1987). However, such spatial associations are not proof of immiscibility, as they merely imply the existence of separate magmas whose intrusion might or might not be coeval as noted by Bowen (Reference Bowen1928). The presumed ‘bimodality’ is actually fallacious and results from rigid adherence to otiose IUGS nomenclature (Le Maitre, Reference Le Maitre2002), which restricts the use of the name ‘carbonatite’ to those rocks containing more than 50 modal per cent primary carbonate and less than 10 wt.% silica. The failings of this nomenclature with respect to carbonatite petrogenesis have been addressed by Mitchell (Reference Mitchell2005b), Gittins et al. (Reference Gittins, Harmer and Barker2005), Tappe et al. (Reference Tappe, Stracke, van Acken, Strauss and Luguet2020), Ackerman et al. (Reference Ackerman, Rappich, Polak, Magna, McClemore, Pour and Čejkova2021) and Mitchell and Gittins (Reference Mitchell and Gittins2022). Curiously, rocks that provide direct textural and mineralogical evidence of gradational relationships between silicate and carbonate rocks, such as calcite pyroxenites and pyroxene calcite carbonatites, are ignored or downplayed in importance. Many of the arguments against transitional rocks presented by Le Bas (Reference Le Bas, Fitton and Upton1987) also fail to recognize the different erosional levels and poor exposure of many carbonatite complexes. As suggested above, episodic magmatism and filter pressing can explain many of the observed geological relationships.
The claim of consanguineous liquid immiscibility is a similar, and commonly unwarranted, assumption that ignores how modern methods of geochronology have the resolution to determine the age of individual units of plutonic–volcanic complexes. For example, Scarrow et al. (Reference Scarrow, Chamberlain, Montero, Horstwood, Kimura, Tamura, Chang and Barclay2022) elucidated the complex temporal magmatic history of the alkaline Miocene Oki-Dōzen volcano (Japan) using zircon U–Th–Pb geochronology. This work showed that pluton formation occurred over a period of 6.4–5.2 Ma by amalgamation and crystallization of discrete pulses of magma.
With respect to carbonatite complexes, Ghabadi et al. (Reference Ghabadi, Brey, Gerdes, Höfer and Keller2022) have determined very accurate U–Pb ages for accessory minerals from individual members of the Miocene Kaiserstuhl complex (Germany) and shown that the main silicate rock types were emplaced over a time span of 1.6 Ma and almost one million years before the carbonatites. Similarly, Weng et al. (Reference Weng, Yang, Niu, Li, Mitchell, Zurevinski and Wu2022) have determined that bastnäsite crystallization from syenite and the younger associated carbothermal veins of the Maoniuping giant REE deposit occurred continuously over a period of 2.5 Ma. Such studies effectively rule out immiscibility for the genesis of both complexes. A further unjustified claim that ‘there was no significant gap between the emplacement of the silicate rocks and carbonatites’ was made by Halama et al. (Reference Halama, Vennemann, Sebel and Markl2005) with respect to the Grønnedal–Íka complex of West Greenland. This hypothesis was based upon a single Rb/Sr isochron age of 1299+/-17 Ma (Blaxland et al. (Reference Blaxland, van Breemen, Emeleus and Anderson1978) established for the rocks of the entire complex. Given the significant error in this age, consanguinity cannot be proved and is of no use for establishing that immiscibility occurred. Similarly, Doroshkevich et al. (Reference Doroshkevich, Ripp and Moore2010) make the unsupportable claim that ‘the close spatial relationships and similar age (130 Ma and 126 Ma respectively) of carbonatites and shonkinite-syenite rocks’ are evidence of liquid immiscibility in the Khaluta complex of West Transbaikalia. Clearly, highly accurate geochronology is now required to interpret the evolution of volcano-plutonic nephelinite–carbonatite complexes such as the archetypal Napak volcano (King Reference King1949), which is known to exhibit a range of Miocene ages (Bishop et al. Reference Bishop, Miller and Fitch1969). Even if contemporaneous magmatism is established by accurate geochronological methods, it offers no proof of immiscibility.
10. Absence of actual geological evidence for liquid immiscibility
There is surprisingly little actual evidence for liquid immiscibility recorded in the textures of alkaline-rock carbonatite complexes. There are certainly no large intrusions or extrusives with the silicate rocks containing large volumes of carbonatite or vice versa, which exhibit macroscopic presumed immiscibility textures. Much of the hypothesis relies on the interpretation of rare small silicate or carbonate ocelli as former immiscible melts. With respect to alkaline rocks, this concept was developed initially by Ferguson and Currie (Reference Ferguson and Currie1971) and Philpotts (Reference Philpotts1976) before the experimental studies described above. Although these initial investigations were concerned primarily with explaining the genesis of co-existing alkaline silicate rocks, Ferguson and Currie (Reference Ferguson and Currie1971) proposed, in a very complex petrogenetic hypothesis involving two periods of immiscibility, that the Callander Bay (Canada) carbonatites were formed from a carbonated olivine nephelinitic magma. With respect to ocelli, Ferguson and Currie (Reference Ferguson and Currie1971, p. 572) stated that the ‘description of these objects as amygdales does not stand examination’, on the basis that the ocelli are sharply bounded and contain minerals identical to those of the matrix. However, this statement applied primarily to the silicate ocelli while the carbonate ocelli were essentially ignored. In fact, a detailed description of the carbonate ocelli was not presented. It was assumed CO2 was exsolved from a carbonated nephelinitic magma after olivine crystallization, which condensed as liquid within a silicate liquid, which then underwent further immiscibility and split into two silicate fractions. How the monomineralic carbonate ocelli are preserved in this process or concentrated to form the Callender Bay carbonatites was not explained. Ferguson and Currie (Reference Ferguson and Currie1971) also conducted experiments on the carbonate ocelli-bearing lamprophyres to test the immiscibility process. However, this was never actually demonstrated and all quenching experiments from 800 to 1100oC at 0.1GPa contained only carbonate-filled vesicles. Unfortunately, the Ferguson and Currie (Reference Ferguson and Currie1971) model was accepted as a feasible means of forming all carbonatites and associated silicate rocks (e.g. Mitchell, Reference Mitchell1980; Andersen, Reference Andersen1988) and its original primary focus on the genesis of alkaline silicate rocks was essentially forgotten. Subsequently, patches of late-forming interstitial calcite in silicate rocks at the Oka and Fen complexes have been interpreted as former immiscible liquid, even though ocelli are not present (Treiman & Essene, Reference Treiman and Essene1985; Andersen, Reference Andersen1988).
Subsequently, the presence of globular structures in the peralkaline nephelinites of Shombole, which instigated the Hamilton–Kjarsgaard experiments, led to further entrenchment of ideas regarding carbonate liquid immiscibility, regardless that carbonate globules are actually extremely rare in volcanic rocks. It has been conveniently forgotten that Peterson (Reference Peterson1989) initially considered the calcite-rich ‘globules’ as vesicles. Unfortunately, we were unable to obtain samples from Shombole, but Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991) have described these globules and found the following minerals in decreasing order of abundance: calcite; Ca–Sr and K–Ba zeolites; fluorite; aegirine; strontianite; fluorapatite; magnetite; sanidine and barium lamprophyllite. In addition, Peterson (Reference Peterson1989, p. 471) stated that unidentified Na–K zeolites can comprise from 0 to 100 vol.% of the globules together with marginal silicate glasses of diverse composition. Textural features, such as the modally-zoned globules, illustrated by Kjarsgaard and Peterson (Reference Kjarsgaard and Peterson1991), especially their Plates 3 and 4, are more suggestive of a vesicular origin rather than an altered former silica-bearing calcitic carbonatite liquid. Clearly, these assemblages have little in common with the calcite spheres formed in the Kjarsgaard and Hamilton (Reference Kjarsgaard and Hamilton1988, Reference Kjarsgaard, Hamilton and Bell1989) experiments. Consequently, their initially proposed equivalence to these is unwarranted.
The rarity of carbonate globules in nephelinite is exemplified in Le Bas (Reference Le Bas, Fitton and Upton1987), who does not describe any immiscibility textures or carbonate globules in nineteen occurrences. Nephelinites, such as those at Napak, apparently contain bona fide vesicles rather than globules (King, Reference King1949). Calcite-bearing globules occur in peralkaline wollastonite nephelinite at Oldoinyo Lengai (this work). We consider this nephelinite to be analogous to the Shombole lavas described by Peterson (1979). The lavas contain many near-spherical globules that do not exhibit quench textures and are filled by discrete crystals of calcite and calcian strontianite (Fig. 4a). Others are broken spheroids with thin veins connecting them to the groundmass, which in many areas, contain irregular voids filled with calcite (Fig. 4b), lobate segregations and pseudomorphs of silicate minerals. However, the continuity of some with the groundmass, and the presence of calcite-filled vugs, suggests that they are more probably either filled vesicles and/or condensates from residual fluids in vesicles.

Figure 4. Carbonate globules and veins in Oldoinyo Lengai wollastonite nephelinite. (a) Complex globules consisting of calcite and/or strontianite with irregular masses and veins of calcite associated with a broken globule. (cross polarized light images); (b) False colour optical image showing carbonate globules and connecting carbonate veins. Nepheline = n; PX = clinopyroxene.
In contrast to nephelinites, melilitites and potassic lavas commonly contain a wide variety of carbonate-rich ‘globules’ and ‘segregations’ e.g. the Cupaello and San Venanzo complexes (Stoppa & Cundari, Reference Stoppa and Cundari1995, Reference Stoppa and Cundari1998) and the Polino carbonatite (Stoppa & Lupini, Reference Stoppa and Lupini1993). Liquid immiscibility has been advocated for all of these examples, and interestingly at Cupaello, the immiscibility of silicate from a carbonate liquid (Stoppa & Cundari, Reference Stoppa and Cundari1995). Globules at Cupaello do not exhibit quench textures and consist of coarse calcite plus minor apatite and magnetite. In common with the Oldoinyo Lengai examples, the globules do not exhibit quench textures, are multiphase and are associated with similar minerals occurring as the filling of spaces in the groundmass.
Carbonate segregations are common in calcite kimberlites such as the Benfontein (Dawson & Hawthorne, Reference Dawson and Hawthorne1973) and Wesselton sills (Mitchell, Reference Mitchell1984). These have never been interpreted to be examples of bona fide liquid immiscibility and are considered to result from in situ differentiation with segregation of a low-temperature carbonate-rich fraction as globules and diapirs due to surface tension effects between deuteric carbonate liquid and co-existing parental silicate liquid. Although the carbonate fraction is immiscible at this stage of the fractional crystallization process and forms discrete bounded globules, these do not result from liquid-liquid immiscibility. Dawson and Hawthorne (Reference Dawson and Hawthorne1973) have noted that such processes, whilst not the result of liquid immiscibility, rise to textures that are identical to those produced by that process. Clearly, a similar origin, in addition to vesicle filling, can account for the carbonate globules in the Oldoinyo Lengai and Italian carbonatites described above.
Recently, Berndt and Klemme (Reference Berndt and Klemme2022) have described a hauyne crystal in the Laacher See (Germany) phonolitic tuff, with nano-to-micron (1–5 microns) inclusions of what are claimed to be quenched carbonate-silicate liquids in the proportions of 4:96%. They illustrate the gradual coalescence of carbonate globules until one of 4 microns diameter is attained. It is argued that these represent the immiscible separation of two contrasted liquids from a phonolite magma. The authors suggest that the hauyne began to crystallise before the onset of immiscibility and that it enclosed minute droplets of the magma. The carbonate blebs contain (wt.%) 15–16 SiO2, 47–52 CaO and ∼ 4 Na2O. These are essentially calcic carbonatites, although the Si most probably represents contributions from the silicate host when analyzed with a 10 micron electron beam spot size. Berndt and Klemme (Reference Berndt and Klemme2022) claim their data support the hypothesis that carbonatites at Laacher See are the products of liquid immiscibility from a phonolite magma. We find this claim to be dubious as there is no textural evidence for this process in the actual intrusive rocks. Supposed examples of immiscibility at Laacher See given by Schmit et al. (Reference Schmid, Wetzel, Cooper, Zou and Wörner2010) appear to be a deformed carbonate clast, interstitial residual carbonate and/or cotectic/eutectic assemblages. Berndt and Klemme (Reference Berndt and Klemme2022) do not provide any information on the modal abundance of the inclusion-bearing hauyne crystals. Further, there is no consideration that the inclusions, which are apparently associated with fractures in the hauyne illustrated by Berndt and Klemme (Reference Berndt and Klemme2022), are merely trapped residual fluids (secondary inclusions), which, although immiscible, are not conjugate. Regardless, the extrapolation of apparently isolated, very rare micron-sized inclusions, as precursors to large volumes of carbonatite seems improbable. For, example, even a 100 micron diameter inclusion with a density of 2.7 requires ∼ 1010 inclusions to form 1 kg of carbonatite.
11. The proposal that as calcite carbonatites have a low Mg content, they cannot have originated from magnesium-rich magmas formed by direct mantle melting
Frequent reference to be found among liquid immiscibility adherents is to calcite carbonatites having a low Mg content; a not surprising observation as it would be miraculous if it were otherwise. From this, it is universally deduced that the parent carbonatite magma, which is never actually defined, could not have originated by direct mantle melting because such magmas are overwhelmingly magnesian and so could not precipitate calcite in abundance. This is a wholly unwarranted assumption. It suffers further from the equally misleading assumption that calcite carbonatites have the same composition as that of the magmas from which they have crystallized. Not only is this not true of most igneous rocks, but it is particularly so for carbonatites where cumulus processes have commonly prevailed during magmatic crystallization.
It is well established that dolomitic magmas can crystallize calcite as long as they have a modicum of alkalies, fluorine and water (Beckett, Reference Beckett1987, Harmer & Gittins, Reference Harmer and Gittins1997, Gittins et al. Reference Gittins, Harmer and Barker2005). In the pseudo-binary join Na2CO3–CaMg(CO3)2 at 0.1 GPa, calcite is the liquidus phase down to a peritectic at 810oC in dolomitic liquids with <23 wt.% Na2CO3. Although this is a peritectic at which, under perfect equilibrium conditions, calcite will disappear by reacting with the residual liquid to form dolomite, there are several factors that limit this reaction. One is that in the experimental system, calcite sinks readily. It is reasonable to suppose that it will do so as readily in natural magmas and lead to the formation of a calcite cumulate, as well as developing disequilibrium conditions. Furthermore, in the experiments, this peritectic reaction is very sluggish and might be similarly so in Nature, further encouraging the uninterrupted crystallization of dolomite. Although thermodynamically permissible, it might be unlikely kinetically, given the sensitivity of the peritectic to changes in temperature and pressure, and a crystallizing liquid is likely to oscillate across the reaction point, resulting in an alternate crystallization of dolomite or calcite. While it is common in igneous petrology to consider equilibrium crystallization, it is highly probable that in carbonatites disequilibrium processes might sometimes be as important, or even more so, than equilibrium crystallization. This is particularly so in the active regime of volcanic complexes. While lubricated by small amounts of the residual liquid, a calcite crystal mush can easily be intruded to form the essentially monomineralic calcite carbonatites, which are common in carbonatite complexes.
The sequence of crystallization, first of calcite followed by dolomite, clearly explains the succession found in so many carbonatite complexes. However, a mistake made all too often by those who assume an original calcite carbonatite magma, formed either by liquid immiscibility or other processes, is the assumption that this sequence will still evolve. This, of course, cannot be as the eventual crystallization of dolomite requires at least a modicum of Mg in the parental liquid.
Another aspect is that the removal of calcite from the evolving system will generate an incomplete (interrupted) reaction at the peritectic and produce calcite overgrown by dolomite, as observed by Bailey and Kearns (Reference Bailey and Kearns2003) in the Kaluwe complex as ‘calcite phenocrysts in a dolomite carbonatite lapillus’ and ‘a dolomite lapillus with coarse calcite in the core’. They considered that these ‘…defy explanation in terms of classic phase diagrams for calcite- dolomite’, but, as we have described above, this is now perfectly explicable. The disequilibrium referred to above also permits the formation of the very common calcite-dolomite carbonatites.
The role of fluorine, given that it is a common constituent of most carbonatite magmas, might be analogous to the effect of Na2CO3 on a dolomitic liquid. Profound lowering of the calcite liquidus temperature by fluorine, introduced as fluorite, was already known from Gittins and Tuttle (Reference Gittins and Tuttle1964), and similar behaviour to that of Na2CO3 is observed in the pseudo-binary system CaF2–CaMg(CO3)2 (Harmer & Gittins, Reference Harmer and Gittins1997) where at 0.1 GPa, calcite is the liquidus phase down to 753o C, which is almost 60oC below the calcite–dolomite peritectic reaction in Na2CO3–CaMg(CO3)2.
The removal of calcite from an evolving dolomitic liquid enriches the residual liquid in Mg and alkalies. However, as most carbonatite magmas contain significant amounts of Si and Al, the accumulating Mg, and some of the Ca, will be abstracted as Fe–Mg silicates while simultaneously increasing the CO2 content. If the solubility limit of CO2 is exceeded, it can be expected to escape from the system, resulting in further limiting the reaction of calcite plus liquid to form dolomite at the peritectic. If the Si and Al are fully consumed in this way, alkali enrichment continues as neither calcite nor dolomite can sequester alkalies.
Additional examples of calcite crystalizing from a magnesian liquid down to about 800o C are provided by the studies of Franz and Wyllie (Reference Franz and Wyllie1966), Fanelli et al. (Reference Fanelli, Cava and Wyllie1985), Otto and Wyllie (Reference Otto and Wyllie1993) and Lee et al. (Reference Lee, Fanelli, Cava and Wyllie2000a, b) of the system CaO–MgO–SiO2–CO2–H2O, in which calcite is found to be a liquidus phase followed at lower temperature by the co-crystallization of calcite and dolomite. The experiments of Franz and Wyllie (Reference Franz and Wyllie1966) and Wyllie (Reference Wyllie and Bell1989) at 0.1 GPa are particularly important as they show that the intersection of a decarbonation reaction with the liquidus permits calcite and forsterite to crystallize together over the temperature range of 970–895o C. The latter temperature is that of the reaction point at which olivine reacts to form monticellite and periclase whilst retaining calcite as a liquidus phase. These phase relationships have determined that at 0.2 GPa, calcite and dolomite can crystallize together from 880 to 650oC, and that liquids with high Mg/Ca ratios will precipitate calcite with very low MgCO3 contents. In addition, Lee and Wyllie (Reference Lee and Wyllie1998), while discussing the vapour (water)-saturated system CaO-MgO-SiO2-CO2, have stated ‘Hydrous magnesiocarbonatite (dolomitic) magmas can precipitate cumulate sövites’.
We conclude that not only it is possible for calcite to crystallize from dolomitic liquids, but also possible for calcite and dolomite to coexist, and even co-crystallize. When calcite sinks into a region rich in earlier precipitated calcite, this creates disequilibrium crystallization of dolomite and produces calcite–dolomite assemblages. Once the peritectic is bypassed, either in equilibrium or disequilibrium conditions, dolomite is the sole carbonate crystallizing and will lead to the formation of dolomite carbonatite.
In rejecting a direct mantle-melting origin of carbonatite magma, most adherents of liquid immiscibility do so on the grounds that mantle-derived magmas are essentially dolomitic. This is an exaggeration as there is a substantial range with compositions intermediate between calcite and dolomite. All the experimentally produced liquids from a variety of mantle peridotite and eclogite compositions have Ca# in the range of 60 to 90 as shown by Wallace and Green (Reference Wallace and Green1998); Thibeault et al. (Reference Thibeault, Edgar and Lloyd1992); Dalton and Wood (Reference Dalton and Wood1993); Sweeney (Reference Sweeney1994); Foley et al. (Reference Foley, Yaxley, Rosenthal, Kisseeva and Jacob2009); Yaxley and Brey (Reference Yaxley and Brey2004); Dasgupta et al. (Reference Dasgupta, Hirschmann and Stalker2006). There are as yet no data on the effect of alkalies on the crystallization of these magmas, but it is not unreasonable to suppose that it would parallel what is already known for Na2CO3 on dolomite.
Intermediate calcite–dolomite magmas clearly exist and can be expected to precipitate magnesian calcite crystallizing as a single-phase carbonate. An example is the Argor complex (Ontario) where carbonates of composition (in mol.%), CaCO3 73.8, MgCO3 21.7, FeCO3 4.0, MnCO3 0.5, are common and crystallized at temperatures in excess of 1000oC. These subsequently exsolved calcite and dolomite as coarse intergrowths with ‘perthitic-like textures’ (Gittins, Reference Gittins1979; Gittins et al. Reference Gittins, Harmer and Barker2005).
Similar carbonates are reported by Yaxley and Brey (Reference Yaxley and Brey2004) in equilibrium with carbonatite liquids formed from the melting of carbonated eclogite at 2.5–5.5 GPa, and by Dalton and Presnall (Reference Dalton and Presnall1998) for carbonates in equilibrium with a model lherzolite. At the Argor carbonatite, there is a further complication; high-temperature carbonates might have crystallized under disequilibrium conditions as they are accompanied by many calcite crystals in which the dolomite content varies considerably between grains which have not exsolved dolomite as lamellae (Gittins et al. Reference Gittins, Harmer and Barker2005).
We conclude that no case can be made for the contention that calcite carbonatites cannot be derived from the mantle by direct melting, or that this establishes liquid immiscibility as the consequent means of genesis. It is well established that calcite can crystallise from a dolomitic mantle-derived melt.
12. Conclusions
We have critically examined the proposition, which has become increasingly popular over the past half-century, that liquid immiscibility is principally responsible for the genesis of calcite and dolomite carbonatites and their associated silicate rocks. After considering the published claims in support of this contention, we have found them wanting. In the Supplementary Material attached to this contribution, we discuss a selection of previously published papers that we believe illustrate many of the misconceptions perpetuated by adherents to the liquid immiscibility hypothesis. Even if a case could be made for liquid immiscibility as explaining the genesis of calcite and dolomite carbonatites, and we do not believe it can, it does not explain the large number of dolomite carbonatites or those calcite carbonatites that are not accompanied by silicate rocks. It is also worth noting that the proponents of liquid immiscibility have advocated an unreasonably wide range of potential parent magma compositions from basanite, nephelinite, phonolite and tephrite to nepheline syenite.
We suggest that liquid immiscibility has, all too often, been invoked without adequate consideration of the validity of data presented in support of the hypothesis (see Supplementary Material). Central to its present appeal are generalizations about spatial and temporal relations; alleged similarities of trace element and isotope compositions and the acceptance of experiments that purport to demonstrate that nephelinite magma evolves to a stage where it splits into two conjugate liquids. We have cast considerable doubt on all of these. We particularly emphasize the error in assuming that because carbonate and silicate liquids are by their nature immiscible, they are consequently conjugate liquids. A further major error is ruling out an origin of carbonatite-forming magma by direct mantle melting on the assumption that such a magma is always dominantly dolomitic and that calcite cannot crystallize from a dolomitic liquid. We have shown that calcite can readily crystallize from dolomite liquid. The assumption also derives from mistakenly presuming that the composition of the carbonatite rocks directly represents that of their parent magma. It is noteworthy that liquid immiscibility is often invoked by uncritical repetition of published assertions that it has already been established. A further fallacy, perpetuated by many petrologists and geochemists, is that experimental studies of the origin of Oldoinyo Lengai nyerereite–gregoryite lavas also explain the genesis of calcite and dolomite carbonatites.
The concept that carbonatites and any associated silicate rocks are generated by liquid immiscibility is a consequence more of its frequent re-telling than of sound experimental and geological considerations. We conclude that no case exists for the genesis of calcitic and dolomitic complexes by liquid immiscibility. Of overwhelming significance is the fact that no experiments have produced dolomite liquid by immiscibility processes, thus eliminating liquid immiscibility for the generation of a very large proportion of the world’s carbonatites. It appears that liquid immiscibility is ‘last resort petrology’.
It is important to note that the objective of this contribution is solely to discredit liquid immiscibility as an explanation for the genesis of carbonatite–alkaline-rock complexes. It is not to provide comprehensive genetic models for all varieties of carbonatites. We seek only to eliminate one of the commonly invoked hypotheses for their genesis. The literature contains many proposals for their origin by partial melting of asthenospheric mantle or partial melting of lithospheric mantle already modified by asthenospheric melts. In both cases, fractional crystallization of the derived carbonate-bearing magmas could lead to carbonatite alone, particularly dolomite carbonatite, or diverse carbonatites with associated silicate rocks. We believe these hypotheses deserve continued assessment.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S001675682300050X
Acknowledgements
This work has been supported by the Natural Sciences and Engineering Council of Canada, Lakehead University, The University of Toronto and Almaz Petrology. Bruce Kjarsgaard is thanked for many comments on his experimental work, although we do not wish to imply that he agrees with our interpretations. Thanks to Sebastian Tappe and Dima Kamenetsky for useful reviews of this paper. Valerie Dennison is thanked for her valuable editorial contributions to the text.







 Open access
Open access