



Healthcare facility-onset Clostridioides difficile infection (HO-CDI), defined as a positive test for C. difficile from an unformed stool specimen on or after hospital day 4, has become increasingly topical due to public reporting requirements and financial penalties for rates greater than expected.Reference McDonald, Gerding and Johnson1 Whole-genome sequencing (WGS) has emerged as an important tool for hospital epidemiology because it can detect single nucleotide polymorphisms (SNPs) among strains.Reference Eyre, Fawley and Rajgopal2 In this report, WGS prompted a focused diagnostic stewardship program (DSP) that has been associated with a significant and sustained decrease in HO-CDI.
Methods
According to standard laboratory protocol, all stool specimens with a provider order for C. difficile testing were processed with a 2-step algorithm, consisting of an initial enzyme-linked immunosorbent assay (ELISA)-based test (C. DIFF QUIK CHEK COMPLETE, Abbott, Chicago, IL) to detect the presence of glutamate dehydrogenase (GDH) and/or toxin, and a DNA amplification test (Illumigene C. difficile, Meridian Bioscience or AmpliVue, Quidel, San Diego, CA), which was run to detect discordant ELISA results. Samples were considered positive if they were GDH and toxin positive or if they were GDH and amplification test positive. The 2-step testing methodology has remained unchanged since 2013, and samples were processed independently of patient demographics.
Between December 2016 and November 2017, stool samples that tested positive for CDI were selected randomly for further analysis. Clostridium difficile was cultured from positive samples, and DNA was extracted and sequencing prepared as previously described.Reference Snesrud, Ong and Corey3 Samples were sequenced using MiSeq Reagent Kit version 3 (600 cycle; 2 × 300 bp; wIllumina, San Diego, CA). Sequencing reads were quality and adapter trimmed and were then assembled de novo using Newbler version 2.7 software. In silico multilocus sequence typing (MLST) was performed using a script designed in house. Basic phylogenetic analyses were performed using Panseq software and initial phylogenetic trees were constructed using Geneious software (Biomatters, Auckland, New Zealand).Reference Laing, Buchanan and Taboada4,Reference Kearse, Moir and Wilson5 Finally, the sequences were filtered for recombination using Gubbins, and a high-resolution SNP-based analysis was performed on clusters of closely related isolates (as determined by Panseq) using an in-house pipeline that utilized Bowtie 2 and SAMtools.Reference Croucher, Page and Connor6
The DSP included provider education regarding WGS results and a weekday review of C. difficile orders placed after hospital day 3 for the following inclusion criteria: >3 stools in 24 hours, the absence of laxative administration, the presence of fever or leukocytosis, or a history of inflammatory bowel disease. An infectious diseases–trained physician and infection preventionists performed the review, aided by an electronic medical record-based testing algorithm that guided appropriate ordering. Stool samples were batch tested each day by the laboratory at 11:00 am, so DSP reviews were conducted early each weekday morning. Samples that did not meet all inclusion criteria were discussed with the primary provider caring for the patient to clarify the indications for testing. Providers were encouraged to cancel orders for samples in which inclusion criteria were not met, but they were allowed to override the recommendations of the DSP team if they still believed testing was indicated. The intervention was instituted throughout the hospital, including the intensive care and oncology units.
We defined HO-CDI as a positive test for CDI from an unformed stool specimen on or after hospital day 4, and we defined community-associated CDI (CA-CDI) as a positive test from an unformed stool specimen prior to hospital day 4. A preintervention versus postintervention cohort analysis assessed the impact of the DSP on the HO-CDI rate. A 12-month baseline period (June 2016–May 2017) was compared with the 12-month intervention period (June 2017–May 2018), and the NHSN calculator was used to compare 2 incidence density rates by exact binomial test.
Results
Molecular epidemiology of C. difficile
WGS was performed on 45 unique patient samples obtained over 12 months, and the median hospital day of collection was day 4 (range, 1–42). In silico analysis of the WGS data assigned the 45 isolates to 21 different multilocus sequence types (MLST), with sequence type (ST) 1 (n = 8 isolates), ST-2 (n = 7 isolates), and ST-42 (n = 6 isolates) being the most common. Phylogenetic analysis clustered the isolates according to MLST (Fig. 1), but a high-resolution SNP-based analysis indicated that even isolates within the same ST differed by multiple SNPs. For example, a high-resolution SNP-based analysis of ST-1 revealed that the isolates were separated by 12–63 SNPs, suggesting that these isolates were related but were distinct strains (Fig. 2A). This conclusion was also supported by a review of the patient data, which could find no epidemiological link among different patients. A similar conclusion was also observed for the ST-2 isolates, where isolates were separated by 5–146 SNPs (Fig. 2B). Notably, 17-267-04463 and 17-367-03811 were separated by just 5 SNPs, which is below the 10 SNP cutoff point suggested by Eyre et al.Reference Eyre, Fawley and Rajgopal2 However, a review of the patient data failed to identify an epidemiological link between the patients. These data suggest that both strains likely emerged from a common ancestor in the past 2–8 years and may be representatives of a ST-2 clone circulating in this geographic region.

Fig. 1. Phylogenetic analysis of Clostridioides difficile isolates. Dendrogram generated using Panseq showing the relationship among 42 of the 45 C difficile isolates. 17-053-03232 (ST-11), 17-056-06956 (ST-11), and 17-086-07498 (ST-37) were removed to aid in visualization because they formed such long branches that they collapsed the tree. Sequences of C. difficile reference strains from NCBI were included for reference (black text). Isolates in red text were classified as hospital onset (HO), and those in green text as community acquired (CA). Traditional sequence types generated in silico from whole-genome sequencing (WGS) data are indicated. The scale bar indicates the approximate number of single-nucleotide polymorphisms (SNPs) per 1,000 base pairs.
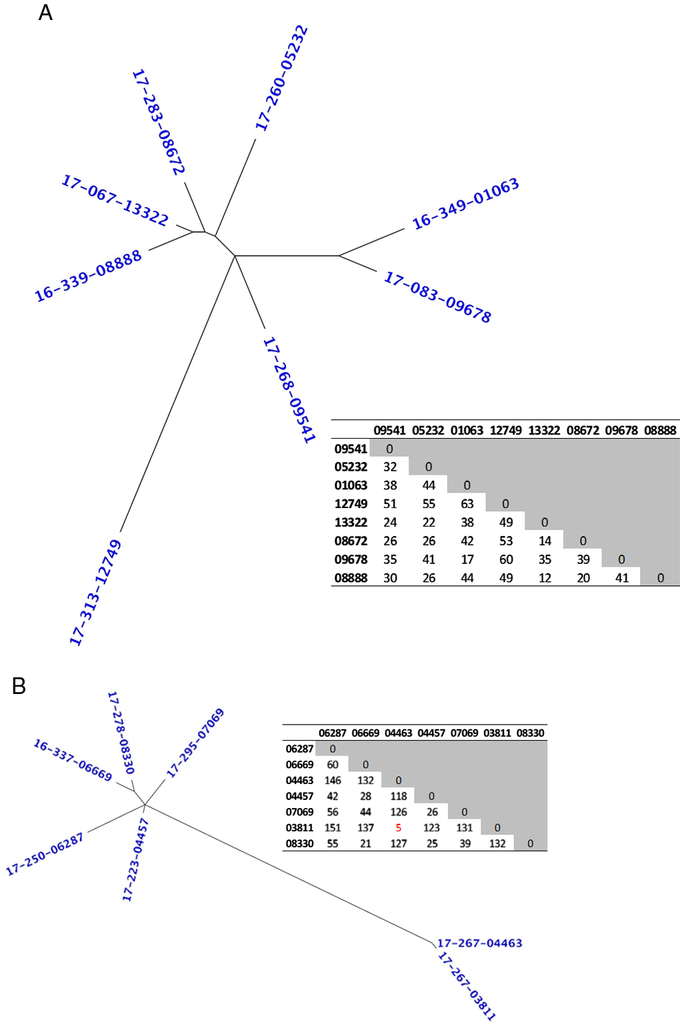
Fig. 2. High-resolution analysis of ST-1 and ST-2 Clostridioides difficile isolates. Unrooted dendrogram and associated single-nucleotide polymorphism (SNP)-based matrix of all isolates from ST-1 (A) and ST-2 (B). The number of high-quality SNPs separating the isolates is displayed in the matrix, and those ≤10 SNPs are highlighted in red text.
Moreover, 17 samples were collected ≤3 days after admission (classified as CA-CDI), and 28 samples were classified as HO-CDI. However, high-resolution SNP-based analysis did not cluster STs into those categories. For example, ST-42 was composed of 2 CA-CDI isolates separated by just 9 SNPs, while all 4 HO-CDI isolates were separated by 19–35 SNPs. Notably, the 2 CA-CDI isolates were cultured 6 months apart from patients located on different floors and with no obvious epidemiological association.
Diagnostic stewardship program
In May 2017, the WGS results from the first 15 C. difficile isolates became available, which reported a lack of clonality. These results prompted the institution of a DSP quality improvement program in June 2017 aimed at providing guidance on testing for C. difficile. A total of 741 tests were ordered, of which 495 (67%) were reviewed. A review was not performed on 246 (33%) tests ordered on weekends or holidays. Indications for testing were met in 251 orders (51%) and were processed from 209 patients, of which 44 tests (21%) were positive. Furthermore, 42 were not submitted for a variety of reasons, including spontaneous cessation of diarrhea, patient discharge, and the identification of an alternative diagnosis. Indications were not present in 244 orders (49%), and 195 orders (80%) were cancelled. For 49 (20%) patients, testing was approved after further discussion with the primary providers, and 9 patients (18.4%) were positive. Also, 2 patients whose tests were initially cancelled experienced continued diarrhea and subsequently tested positive for CDI and responded well to treatment. No cases of severe C. difficile infection occurred among patients whose testing was cancelled. Testing decreased from 135 per 10,000 patient days in the baseline period to 80 per 10,000 patient days in the intervention period (P < .0001). The HO-CDI rate decreased from 11.67 per 10,000 patient days in the 12-month baseline period to 6.25 per 10,000 patient days (P = .0008) in the 12-month intervention period. A nonsignificant change in the rate of CA-CDI was detected: 6.38 per 10,000 patient days in the baseline period and 5.70 per 10,000 patient days in the intervention period (P = .40).
Discussion
Similar to other reports, we identified a genetically diverse population of C. difficile isolates, pointing to a complex epidemiology with no clear pattern of acquisition.Reference Eyre, Fawley and Rajgopal2,Reference Kociolek, Gerding, Espinosa, Patel, Shulman and Ozer7 Although we only sequenced single colonies from each patient and thus may have missed heterogeneity among colonizing strains and cannot exclude transmission from asymptomatic carriers or the environment, the divergence among isolates argues against hospital-based transmission from other symptomatic patients.
Based on the initial report of 15 isolates without clonality, we launched a focused DSP to improve testing. Subsequent WGS batch reports confirmed the initial findings, and although skeptical of the impact of a DSP, our experience was illuminating; we often observed reflex testing for CDI rather than investigations for more likely causes of diarrhea. Since completing data analysis in May 2018, the program has continued. The HO-CDI rate decreased to 3.2 per 10,000 patient days for the last 7 months of 2018. Potential confounding factors to our findings include a global decrease in CDI (both HO-CDI and CA-CDI) unrelated to our DSP, or heightened provider awareness of the definition of HO-CDI, which resulted in more frequent testing prior to hospital day 4 so that positive results would be attributed as CA-CDI rather than HO-CDI. However, the rate of CA-CDI did not change significantly during the evaluation period, which argues against both of those influences.
Previous studies have reported the impact of DSP on HO-CDI rates, but to our knowledge, ours is the first to link it to molecular epidemiology.Reference Quan, Yim and Merrill8,Reference Truong, Gombar and Wilson9 WGS has provided novel data on the transmission patterns for C. difficile, which is, in turn, prompting adjustments on appropriate diagnostic and infection prevention responses. A focused DSP, informed by WGS, allowed for the rapid translation of results from bench to bedside, and future studies will undoubtedly reveal continued expansion of this partnership.
Author ORCIDs
Glenn Wortmann, 0000-0002-7801-8871
Acknowledgments
We acknowledge Ms Katarzyna Magda, MS, MT (ASCP), for microbiology support and Ms Janette Ponticello, RN, and Ms Pam Farrare-Wilmore BS, MT (ASCP), CIC, for infection prevention support. We also thank the staff of the Multi-Drug Resistant Organism Repository and Surveillance Network (MRSN) for their assistance with whole-genome sequencing.
Financial support
No financial support was provided relevant to this article.
Conflicts of interest
The authors have no conflicts of interest to report, and no funding was received to conduct this study.

