



Introduction
Atomic force microscopy (AFM) is a powerful tool that allows the comprehensive study of mechanical properties and interactions with nanometer resolution. The last three decades have established the technique as the instrument of choice for high-end structural analysis of samples ranging from single molecules to complex biological systems, such as proteins and cells. Unlike other high-resolution imaging techniques, such as super-resolution light and advanced electron microscopy, AFM does not require any sample labeling or modification. In turn, that means that no preparation artifacts are introduced during imaging, thus enabling the capacity to study the specimens at their near-physiological or native state/environment.
High-resolution imaging techniques, such as X-ray crystallography, transmission electron microscopy, as well as conventional AFM, have contributed to our in-depth understanding of various biological systems. Nevertheless, they have struggled with the functional interpretation of molecular and dynamic processes taking place in living systems, by only partially addressing temporal snapshots of specific biological events and processes. Studying single macromolecule dynamics and the function of complex biological systems, such as individual living cells, requires a tool that can provide both high spatial and high temporal resolution.
High-speed AFM has come a long way since the first attempts to visualize biomolecular dynamics [Reference Drake1], or since the first postulated theoretical considerations for the scan speed limitations in conventional AFM [Reference Butt2]. Developments in the last 15–20 years have made possible the application of ultra-short cantilevers, high-resonance piezoactuator-based sample scanners, feedback systems, and optical beam deflection detectors that have ultimately enabled the studies of dynamic biomolecular processes with high-speed prototypes [Reference Viani3,Reference Ando4], as well as commercial systems enabling both tip [Reference Stamov5,6] as well as sample-scanning acquisition rates of 10–20 frames per second [Reference Ando7]. This has effectively been made possible by the much higher oscillation frequencies of the used cantilevers, improved feedback bandwidths, as well as enhanced XY-movement, which in turn enable higher scanning rates without decreasing the resolution of the measurement, or damaging very sensitive samples, as reviewed in [Reference Ando8,Reference Amrein and Stamov9].
We have recently developed and launched the NanoRacer®, which is currently the fastest commercial high-speed AFM, able to operate at a video-rate scanning speed of 50 frames/sec. The instrument features 3-axis closed-loop sample scanning, high detector bandwidth of 8 MHz, novel high-speed power amplifier, as well as advanced algorithms for scanner control and feedback loop error correction, such as iterative inversion-based adaptive scanning and harmonic motion to improve feedback loop bandwidth [Reference Lyman10]. This is further complemented by the use of dynamic proportional-integral-derivative (PID) control, minimizing feedback saturation errors. In combination with a small excitation laser spot size, this enables the high-speed stable successive imaging of soft and fragile biological specimens with a temporal resolution of near-20 ms. The NanoRacer was a recipient of the Microscopy Today 2021 Innovation Award based on its usefulness to the microscopy community [Reference Lyman10].
In this article we demonstrate how high-speed AFM can be applied for studying single-molecule dynamics in biological systems, for example, studying lipid ripple phase formation, thermodynamic DNA rehybridization, collagen type I fibril formation, as well as annexin V (A5) rotational dynamics.
Methods and Materials
Sample preparation
All measurements were carried out in a customized fluid chamber and optional temperature control including a muscovite mica disk as a support. To expose the atomically flat mica surface, the substrates were freshly cleaved prior to sample preparation.
DMPC lipid bilayers
150 μM 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) solutions were prepared by dissolving DMPC (Avanti Lipids) in sample buffer (150 mM NaCl, 20 mM Tris, 5 mM EDTA buffer (pH 7.6)). The protocol included depositing 5 μL of DMPC solution to the substrate, adding 1 μL of 100 mM CaCl2, followed by 45 min incubation at 4°C. After 2-fold rinsing in sample buffer, the specimens were immediately imaged.
DNA origami nanostructures (DONs).
DONs were provided by collaborators [Reference Domínguez11] and prepared as previously described [Reference Angelin12]. Each rectangular origami template carried five covalently bound biotin residues. 10 μL of 1 nM of DNA origami solution in TE-1x buffer (40 mM Tris, 2 mM EDTA, 12.5 mM Mg2+, pH 8) were incubated on clean substrates for 3 min. The fluid cell was topped with 1.5 mL TE-1x buffer, followed by adding 10 μL of 1 μM streptavidin (STV) stock (Sigma-Aldrich).
Plasmid DNA.
Freshly cleaved mica substrates were coated with 10 μl of 0.01% 30-70 kD poly-L-ornithine stock solution (Sigma-Aldrich) for 10 min. After rinsing with ultra-pure water, 10 μL of pUC19 vector solution (MoBiTec) was deposited on the surface, to reach a final concentration of 1 nM DNA in the chamber volume. After another rinsing step with ultra-pure water, the substrates were subjected to imaging in fluid.
Collagen type I nanomatrices
After being freshly cleaved, the mica substrates were incubated with PBS-1x buffer (200 mM KCl, pH 7.3) for 30 min. After adding 15 μL of type I bovine atelocollagen stock solution (Advanced Biomatrix) to a final volume collagen concentration of 30 μg/ml, the samples were immediately subjected to imaging in PBS-1x buffer.
Annexin V lattices
Coagulation solutions were prepared from Coag Reagent II, containing DOPC:DOPS (7:3) (Avanti Lipids). Briefly, 10 μL of Coag Reagent II were deposited in clean Eppendorf tubes and vacuum-dried for 3 hr, before being diluted in 1 mL coagulation buffer (20 mM HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7.4), and ultra-sonicated for 30 min. 10 μM A5 aliquots were prepared from 33 kD human placental A5 (Sigma-Aldrich) in A5 buffer (20 mM Tris-HCl, 150 mM NaCl, pH 7.6). After freshly cleaving the mica substrates, 10 μL of Coag solution was deposited for 2 min and rinsed with Coag buffer. The fluid cell was topped with 1 ml of Coag buffer, and the A5 concentration was adjusted to 100 mM.
High-speed AFM imaging
Samples were analyzed using a NanoRacer® High-Speed AFM (Bruker), which features a customized fluid cell with optional temperature control. The instrument was placed in an acoustic isolation housing on an active antivibration table. Unless stated otherwise, all samples were measured in liquid under ambient temperature in amplitude-modulation AC mode. We used fast-scanning high-resonant ultra-short cantilevers (USC-F1.2-k0.15, NanoWorld, Switzerland) with a nominal resonance frequency of 1.2 MHz in air, spring constant of 0.15 N/m, reflective chromium/gold-coated silicon chip, and high-density carbon tips with a radius of curvature smaller than 10 nm.
Results
Temperature-induced lipid phase transition
The temperature-driven increase in mobility between solid and fluid phases in supported lipid bilayers often results in intermediate ripple-like structures, where both states coexist with a constant periodicity [Reference Takahashi13]. To study the dynamics of that process in detail we have imaged supported DMPC lipid bilayers in the temperature range from 22°C to 24°C (Figure 1).
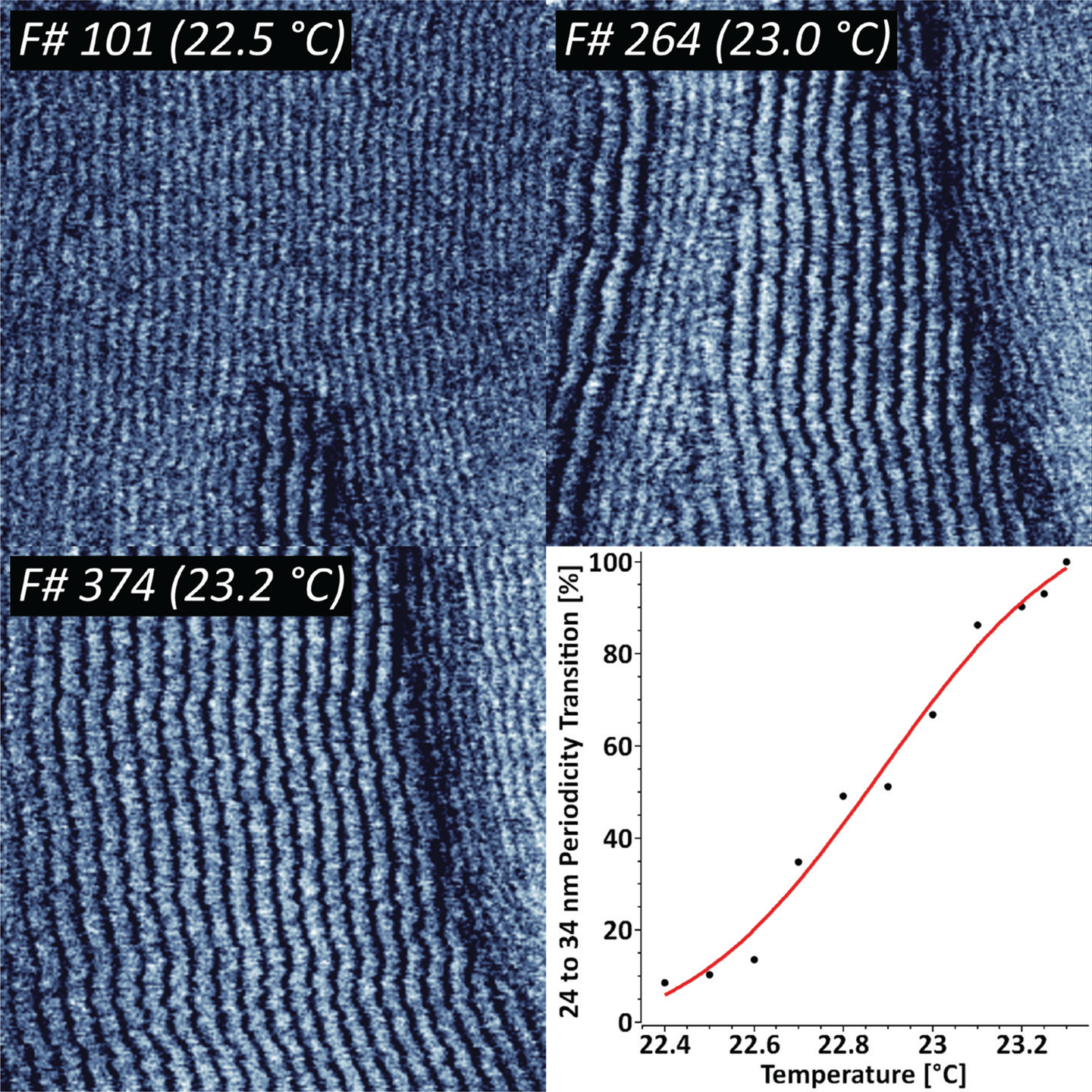
Figure 1: Sequence of 424 AFM phase images recorded at 1 frame/sec, indicating the thermodynamic transition between two distinct DMPC ripple phase periodicities. The ripple phase transition kinetics has been fit with a Poisson-Boltzmann equation. XYZ-scales in the AFM images are 800 nm, 800 nm, and 3 deg respectively.
The results indicate that there are two concurring structural periodicities existing in the studied temperature range, namely 24 and 34 nm. The gradual temperature ramping from 22.4°C to 23.3°C leads to a complete 34 nm ripple phase transition. Further increase in temperature above 25–26°C led to a complete loss in periodicity. For representative purposes, the periodic transition has been fit with a Poisson-Boltzmann theory, which is typically applied for model membranes with mobile charged lipids [Reference Fleck14].
DNA origami streptavidin-biotin binding kinetics
DNA origami nanostructures (DONs) are emerging as molecular pegboards for the immobilization of ligands on surfaces, often featuring high-affinity biotin-streptavidin bridges. We have studied DONs that carry five equidistantly spaced covalent biotin tags (Figure 2).
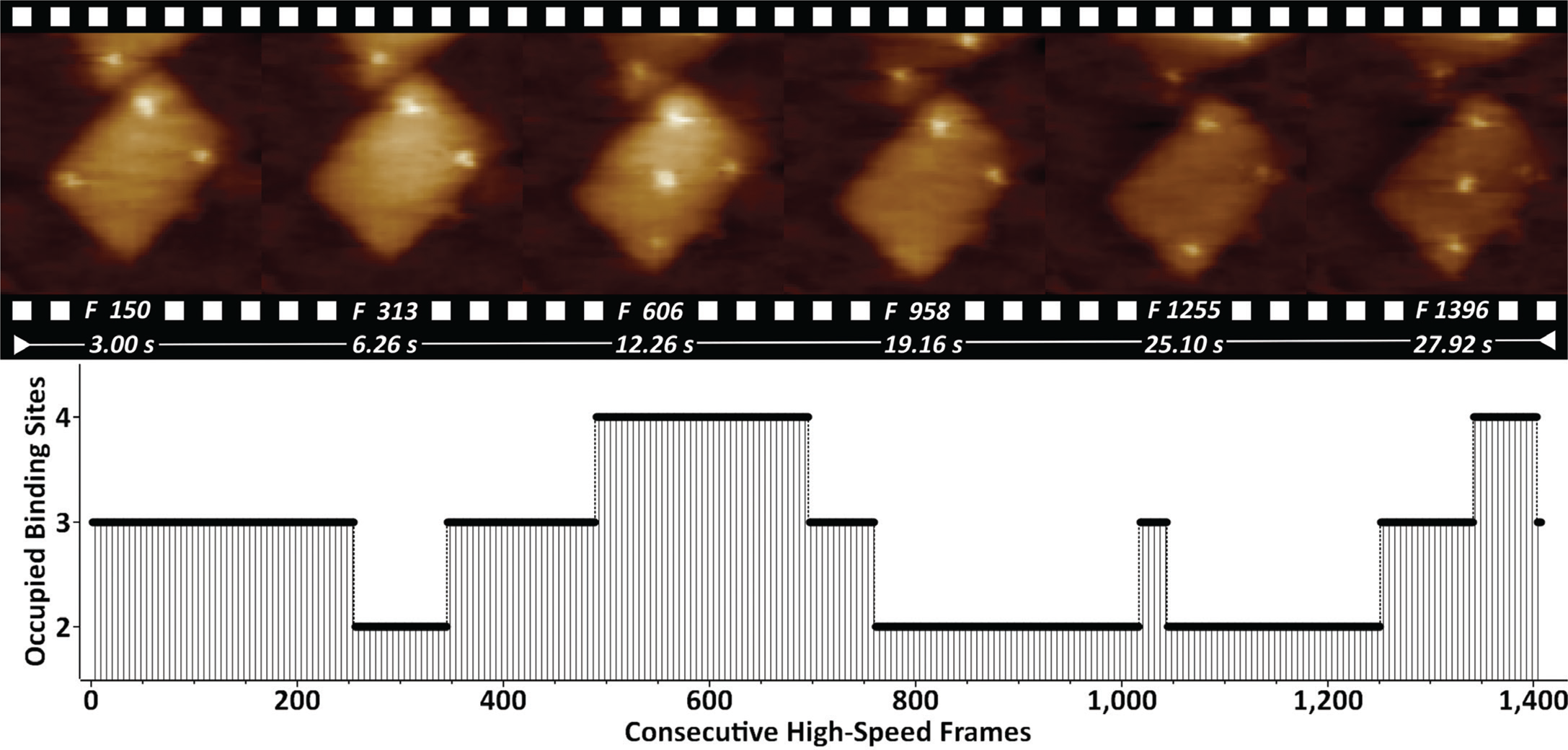
Figure 2: Biotinylated DONs imaged in buffer in the presence of streptavidin. Streptavidin binding/unbinding is seen on top of the DONs as constantly appearing/disappearing bright dots. The entire 1407-frame high-speed video sequence was recorded with 50 frames/sec. Site-occupation analysis is shown on the bottom. XYZ-scaling in the AFM images is 150, 150, and 2 nm respectively.
By supplementing the biotinylated DONs with streptavidin, it is possible to visualize the dynamics of binding/unbinding of individual streptavidin molecules. Quantification of the binding properties by occupation site analysis showed that the most occupied states featured 2 (39%) and 3 (41%) binding sites.
Thermodynamic rehybridization of DNA.
We have previously shown that pUC19 plasmids bind to polycationic interfaces in the form of structures that carry a lot of torsional energy, in turn leading to a higher propensity of forming dehybridization bubbles [6]. By subjecting that system to high-speed AFM imaging at 40 frames/sec, we could visualize the thermodynamic state fluctuations of the single DNA strands [Reference Manghi and Destainville15] and ultimately their rehybridization to a double-stranded state (Figure 3).
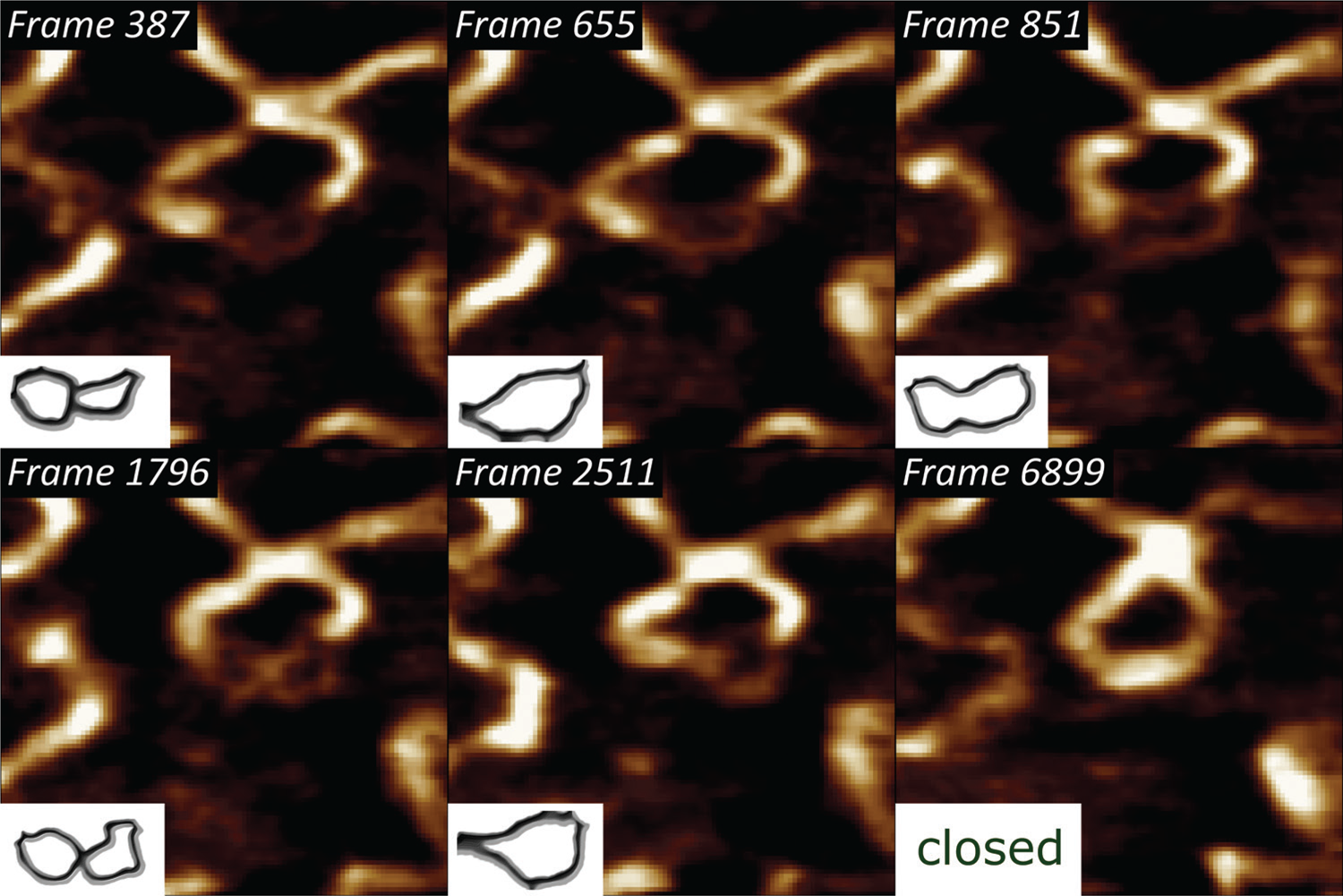
Figure 3: Selected frames from a 6915-frame-long high-speed DNA bubble rehybridization sequence, showing individual single-stranded DNA conformation states, imaged with a temporal resolution of 25 ms. The lower left insets indicate the backtraced shapes of the metastable DNA bubble. XY-scaling of the amplitude AFM images is 100×100 nm2.
Kinetics of collagen type I fibrillogenesis
Collagen type I is the most abundant extracellular matrix protein in mammalian cells. We have previously reported on the application of fast-scanning AFM for studying the epitaxially driven hierarchical self-assembly of collagen type I fibrillar films [Reference Stamov16]. Here we have further demonstrated that the kinetics of the fibrillogenesis can be studied with up to 15 frames/sec (Figure 4).
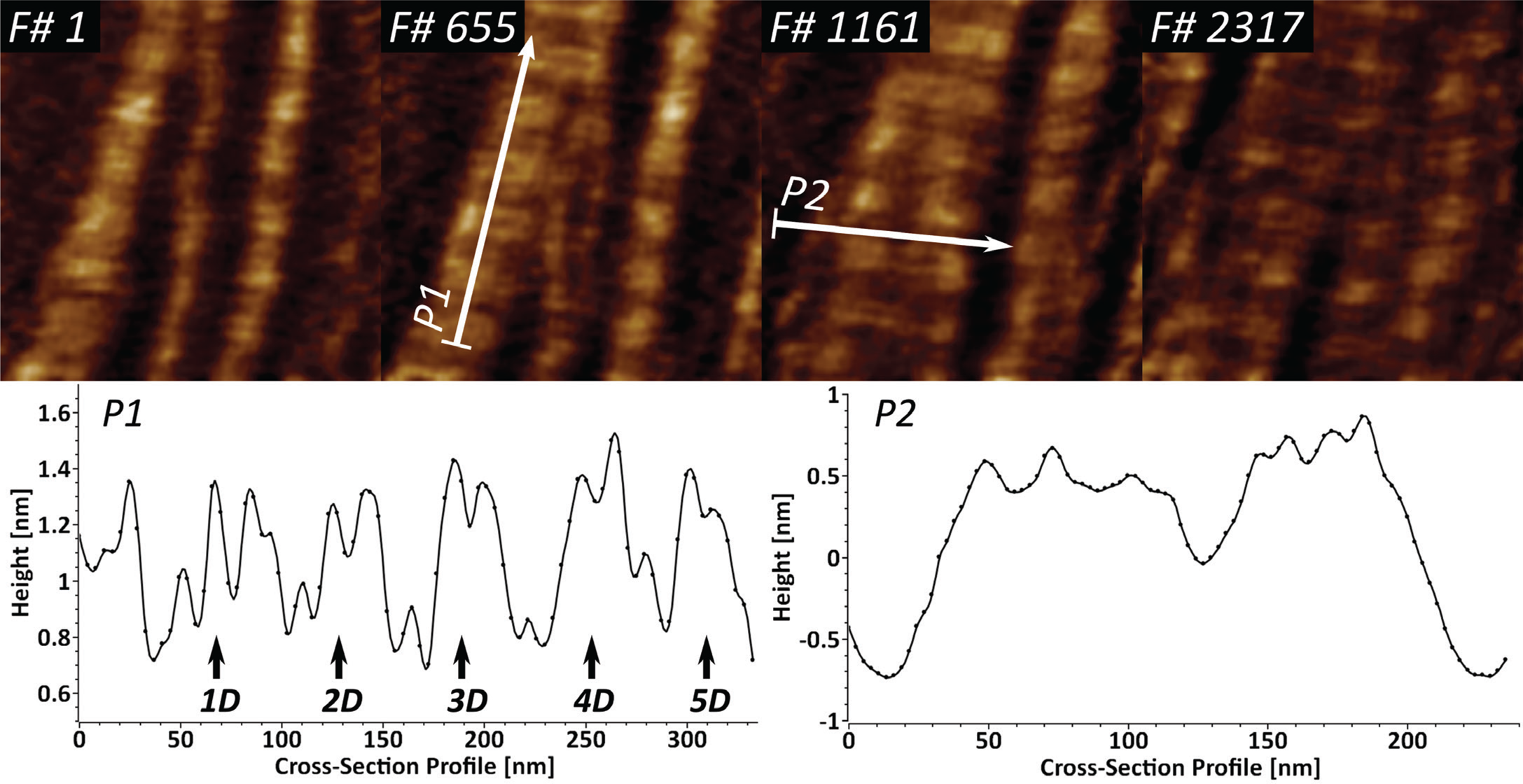
Figure 4: High-speed imaging of collagen type I fibril formation at 15 frames/sec, studied over near 3900 frames. Cross-section profile P1 identifies the repeating characteristic D-band (67 nm) and shows the existing sub-D-banding structure. Profile P2 is used for identification of several speculative microfibrillar units in the fibrillar films.
Upon closer investigation of the cross-section profile P1, it becomes apparent that the D-banding periodicity of collagen I consists of several sub-D-bands, which have been speculated to be a result of lateral amino acid staggering. The cross-section profile P2 shows that it is possible to identify several peaks with a lateral dimension of 5–8 nm, which appear to be structural intermediates in the formation of the larger collagen fibrils.
Rotational dynamics of mobile annexin V trimmers
Annexin V (A5) serves as an important regulator of membrane repair in eukaryotic cells, where it shows a strong Ca2+ binding affinity to phosphatidylserine [Reference Bouter17,Reference Gauer18]. We have used high-speed AFM to study the 2D crystal formation in a model system containing supported lipid bilayers and trimeric A5 molecules, forming a stable honeycomb P6-symmetry lattice. We demonstrate the lateral dynamics of the mobile A5 trimers and show how the A5 structure orientation can be resolved by acquiring multiple high-speed AFM images (Figure 5).
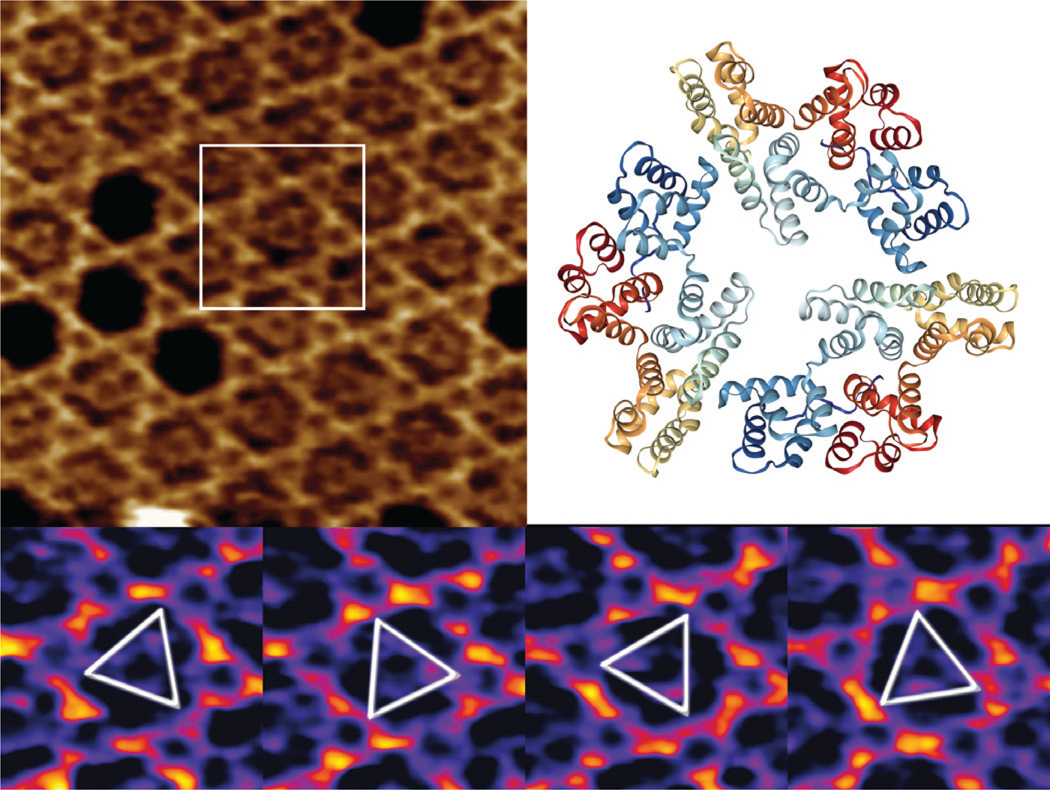
Figure 5: High-speed AFM imaging of an A5 P6 honeycomb lattice, with central non-P6 positions occupied by mobile A5 trimers (top left). A 3D view of a “2IE7” A5 PDB trimer (from www.rcsb.org) with a global C3 symmetry (top right) is given for comparison only. Outlined inset is shown in the bottom panel at temporal resolution of 833 ms, identifying several preferred orientations of the rotational A5 dynamics. XYZ-scales in top left and bottom panels are 100×100×1.5 nm3 and 32×32×1.5 nm3 respectively.
While two-thirds of the trimers occupy the A5 P6-lattice, the mobile A5 fraction has very high rotational dynamics, in which it intermittently interacts with the honeycomb lattice in preferred orientations at 0° and 60°.
Discussion
Under certain experimental conditions, supported lipid bilayers adopt different states, namely solid (gel) and fluid (liquid-ordered and liquid-disordered) phases. The occurrence is specific to the chemical nature of the lipids, and transition from solid to fluid phase is related to a positive temperature shift. Previous studies have suggested that increase in temperature can include transition between several ripple states, namely dominant and metastable phases with different periodicities [Reference Takahashi13]. Our results indicate that this process can be quantified by means of high-speed AFM, by following the complete 24 to 34 nm periodicity transition while heating the sample from 22.4°C to 23.3°C (Figure 1). The complete loss of periodicity is consistent with the full transition to a fluid state above 26°C.
The bottom-up self-assembly of DONs typically features extra molecules that can trigger cell receptor stimulation [Reference Hu19] and early signaling events [Reference Lanzerstorfer20]. The monitored dynamics of biotin-streptavidin binding in such a scenario (Figure 2) indicates that further molecular tailoring can be successfully exploited by featuring enzymes, growth factors, or additional epitopes. Currently we are working on fine-tuning the streptavidin-biotin binding by using enhanced mono- and bivalent streptavidin protocols [Reference Domínguez11].
Supercoiled DNA states, characteristic for interactions with molecules, such as other nucleic acids, DNA-specific enzymes, etc., can drive partial “unzipping” of the double-stranded DNA regions [Reference Jeon21]. It is generally driven by a torsional stress around the strand axis. Once the molecule is dehybridized, and due to the hydrophobic organic base backbone, the single strands twist around the axis to minimize contact with water (Figure 3).
Several X-ray structural models predicted that the very hierarchical structure of collagen type I involves different structural intermediates, one of which is a microfibrillar unit (4–5 nm) that ultimately gives rise to thicker fibrillar structures with a characteristic D-banding of 67 nm [Reference Perumal22,Reference Orgel23]. Our results show that the dynamics of collagen type I fibrillogenesis can be studied successfully with high-speed AFM, while also indicating the existence of several structural intermediates in the fibril formation (Figure 4). The identified 5–8 nm wide fibrillar units, after accounting for certain AFM tip convolution, can be attributed to the already-mentioned microfibrillar units. The sub-D-banding periodicity, in combination with the high temporal resolution, here can be applied as a molecular fingerprinting tool for studying collagen type I mutations in lab diagnostics.
The high-speed AFM imaging of mobile A5 trimers within the stationary P6 lattice indicate that preferred structural orientations at 60° can be studied (Figure 5), but realistically the rotational dynamics of the process is at least a few orders of magnitude faster than the high-speed AFM imaging rates. As suggested previously [Reference Heath and Scheuring24], further studies of A5 rotation kinetics would require the application of single line, or even point scanning, to boost the temporal resolution.
Conclusion
This article describes the application of high-speed AFM for studying several biological systems with acquisition rates of up to 50 frames/sec. With no requirement for sample processing, high-speed AFM enables measurements of samples at their near-native state. With acquisition line rates of up to 5 kHz, the high-speed AFM used here offers a 3-fold boost in temporal resolution compared to conventional AFM. In turn, this enables the real-time studies of dynamic and molecular interactions such as single molecule binding dynamics, tracking of protein-protein and protein-DNA interactions, DNA rehybridization dynamics, monitoring of enzyme kinetics, lipid remodeling in multi-component membranes, etc. Current studies are underway to demonstrate how high-speed force spectroscopy applications, including nanomechanical mapping of single molecules, membrane segregation, and novel unfolding pathways of biomolecules in health and disease would additionally complement our knowledge of the molecular dynamics in both life science and material science applications.
Acknowledgements
We would like to thank Carmen M. Domínguez and Christof M. Niemeyer (KIT, Karlsruhe, Germany) for providing the DON samples used in Figure 2.





