



Introduction
Angiostrongylus Kamensky, 1905 or the lungworm is a parasitic nematode in the superfamily Metastrongyloidea (Spratt, Reference Spratt2015). To date, over 20 species of this genus have been reported around the world, and Angiostrongylus cantonensis and Angiostrongylus costaricensis have been reported to be causative agents of neurological and abdominal angiostrongyliasis in humans, respectively (Spratt, Reference Spratt2015; Barratt et al., Reference Barratt, Chan, Sandaradura, Malik, Spielman, Lee, Marriott, Harkness, Ellis and Stark2016). Another species found in Asian countries, Angiostrongylus malaysiensis, is also a potential human pathogenic parasite (Ansdell and Wattanagoon, Reference Ansdell and Wattanagoon2018). In Thailand, A. cantonensis and A. malaysiensis are the most common species (Watthanakulpanich et al., Reference Watthanakulpanich, Jakkul, Chanapromma, Ketboonlue, Dekumyoy, Lv, Chan, Thaenkham and Chaisiri2021). Angiostrongylus cantonensis is a well-known pathogen that causes oeosinophilic meningitis associated with angiostrongyliasis in humans in Thailand, whereas A. malaysiensis is increasingly reported in many provinces in the country (Eamsobhana, Reference Eamsobhana2013; Watthanakulpanich et al., Reference Watthanakulpanich, Jakkul, Chanapromma, Ketboonlue, Dekumyoy, Lv, Chan, Thaenkham and Chaisiri2021).
Angiostrongylus cantonensis and A. malaysiensis are similar in terms of life cycle, host range, host habitat and morphological and genetic information (Bhaibulaya, Reference Bhaibulaya and Cross1979; Eamsobhana et al., Reference Eamsobhana, Lim and Yong2015; Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020). These 2 species of Angiostrongylus can infect the same species of definitive and intermediate hosts (Bhaibulaya and Techasoponmani, Reference Bhaibulaya and Techasoponmani1972). In addition, mixed infections of A. cantonensis and A. malaysiensis in snail intermediate hosts and rodent definitive hosts have been widely reported (Eamsobhana et al., Reference Eamsobhana, Yong, Prasartvit, Wanachiwanawin and Boonyong2016; Watthanakulpanich et al., Reference Watthanakulpanich, Jakkul, Chanapromma, Ketboonlue, Dekumyoy, Lv, Chan, Thaenkham and Chaisiri2021). This may lead to difficulty in discriminating between A. cantonensis and A. malaysiensis. Adults of A. cantonensis and A. malaysiensis can be morphologically differentiated by the minute protrusion at the posterior end of females and the bursal rays of males (Bhaibulaya, Reference Bhaibulaya and Cross1979; Thiengo et al., Reference Thiengo, Maldonado, Mota, Torres, Caldeira, Carvalho, Oliveira, Simões, Fernandez and Lanfredi2010). Nevertheless, the morphological variance between the 2 species can confound identification. Moreover, differences between the morphological characteristics of the larval stages, especially the infective stage, have not yet been clarified. Therefore, the identification of Angiostrongylus species based on morphological characters is difficult due to vague and similar descriptions of size and body shapes among species (Robles et al., Reference Robles, Navone and Kinsella2008; Monte et al., Reference Monte, Gentile, Garcia, Mota, Santos and Maldonado Júnior2014).
Recently, molecular analysis has been used to differentiate various Angiostrongylus species (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020; Anettová et al., Reference Anettová, Izquierdo-Rodriguez, Foronda, Baláž, Novotný and Modrý2022). Molecular and phylogenetic studies for discriminating closely related Angiostrongylus spp. have used mitochondrial and nuclear genes as genetic markers; the mitochondrial genes include the cytochrome c oxidase subunit I (COI), cytochrome b (cytb), 12S rRNA and 16S rRNA genes (Rodpai et al., Reference Rodpai, Intapan, Thanchomnang, Sanpool, Sadaow, Laymanivong, Aung, Phosuk, Lammaunwai and Maleewong2016; Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019; Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020), and the nuclear genes include the 66-kDa protein gene, small subunit ribosomal RNA (18S rRNA) gene and internal transcribed spacer 2 (ITS2) region (Eamsobhana et al., Reference Eamsobhana, Lim, Zhang, Gan and Yong2010, Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019; Rodpai et al., Reference Rodpai, Intapan, Thanchomnang, Sanpool, Sadaow, Laymanivong, Aung, Phosuk, Lammaunwai and Maleewong2016; Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019). Furthermore, the whole mitochondrial genome has been employed for phylogenetic analysis and species distinction (Valentyne et al., Reference Valentyne, Spratt, Aghazadeh, Jones and Šlapeta2020).
Molecular phylogeography analysis of A. cantonensis and A. malaysiensis may provide insight into specific genetic variation and population formation (Avise, Reference Avise2000). The phylogeography based on COI sequences of A. cantonensis from Thailand, Taiwan, China and Japan revealed that the geographical distribution of A. cantonensis probably reflects multiple independent origins that were likely to have been influenced by human activities (Tokiwa et al., Reference Tokiwa, Harunari, Tanikawa, Komatsu, Koizumi, Tung, Suzuki, Kadosaka, Takada, Kumagi, Akao and Ohta2012). Likewise, Monte et al. (Reference Monte, Simões, Oliveira, Novaes, Thiengo, Silva, Estrela and Maldonado2012) analysed the phylogenetic relationship of COI sequences for A. cantonensis and revealed that some haplotypes from Brazil clustered with isolates from Asia, while the rest formed distinctly divergent clades, indicating multiple origins of A. cantonensis in Brazil. In addition, the phylogeny based on the 66-kDa protein gene revealed distinct clades among A. costaricensis, A. cantonensis and A. malaysiensis. However, no clear separation of the conspecific taxa between A. cantonensis and A. malaysiensis from different geographical regions was reported. Greater sample sizes of the conspecific taxa from each locality may provide a conclusive inference of distinct phylogeographic patterns (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). Moreover, there have been no reports on the molecular identification of third-stage larvae of A. cantonensis and A. malaysiensis using the 66-kDa protein gene. Therefore, we further analysed the genetic diversity of A. cantonensis and A. malaysiensis in Thailand using 66-kDa protein gene sequences. The phylogeny and haplotype network of a 66-kDa protein gene in A. cantonensis and A. malaysiensis were also analysed to determine the relationship between hosts and parasites.
Materials and methods
Angiostrongylus worms
The total 55 Angiostrongylus samples consisted of 14 specimens of A. cantonensis and 41 specimens of A. malaysiensis. Within the 14 specimens of A. cantonensis, 2 female worms were collected from a definitive rodent host (Bandicota sp.; n = 1) in Kamphaeng Phet province, central Thailand, and 12 third-stage larvae (L3) were previously collected from an intermediate land snail host (Achatina fulica; n = 30) from Chaiyaphum province, northeastern Thailand (Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019). Of the 41 A. malaysiensis samples, 38 L3 specimens were collected from 21 A. fulica in Chiang Rai province, and the remaining 3 L3 specimens were collected from 1 A. fulica in Phrae province in northern Thailand (Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019), as shown in Fig. 1. Adult worms were fixed in absolute alcohol and stored at −20°C until DNA extraction. The genomic DNA samples of Angiostrongylus larvae were stored at −20°C.

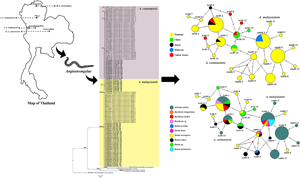
Fig. 1. Map of Thailand showing the number of samples of representative Angiostrongylus spp. The red and black circles indicate Angiostrongylus cantonensis and Angiostrongylus malaysiensis, respectively.
DNA extraction
Before performing the DNA extraction, adult worms were dried by placing on filter paper for 15 min at room temperature. Individual worms were excised into small pieces, which were then placed in 1.5 mL microcentrifuge tubes, crushed and digested in lysis buffer. Genomic DNA extraction was performed using the NucleoSpin® Tissue kit (Macherey-Nagel, Duren, Germany) according to the manufacturer's protocol. Genomic DNA samples of Angiostrongylus larvae that had been collected previously (Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019) from A. fulica were used as samples in this study. The genomic DNA was checked by running it on a 0.8% agarose gel in 1× TBE buffer at 100 V. The gel was stained with ethidium bromide, destained with distilled water and photographed under ultraviolet light. The DNA solution was stored at −20°C prior to further processing.
Polymerase chain reaction (PCR) and sequencing
The DNA fragment (300 bp) of the 66-kDa protein gene was amplified using primers AC1 5′-CTCGGCTTAATCTTTGCGAC-3′ and AC2 5′-AACGAGCGGCAGTAGAAAAA-3′ (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). The PCR components (30 μL final reaction volume) contained 15 μL of EconoTaq® PLUS 2× Master mix (1×; Lucigen Corporation, Middleton, WI, USA), 1.5 μL of each primer at 5 μ m (0.25 μ m), 9 μL of distilled water and 3 μL of the DNA template (20–200 ng). The PCR cycle was initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 2 min, annealing at 58°C for 1 min and extension at 72°C for 3 min, with a final extension at 72°C for 7 min. All PCRs were performed in a Biometra TOne Thermal Cycler (Analytik Jena AG, Jena, Germany). The amplified products were analysed using 1.2% agarose gel electrophoresis. Purification of the PCR products was performed using a NucleoSpin® Gel and PCR Clean-Up Kit (Macherey-Nagel, Germany) following the manufacturer's instructions. The purified PCR products were run on a 1.2% agarose gel at 100 V in 1× TBE buffer. The PCR products were sequenced in both the forward and reverse directions at Macrogen Inc., Seoul, Korea.
66-kDa protein sequences from GenBank
The nucleotide sequences of a 66-kDa protein-encoding gene of A. cantonensis and A. malaysiensis from Thailand, China, Japan, Malaysia and the United States downloaded from GenBank were included in the present study (Table S1). In addition, the sequences from Ancylostoma caninum and Heterorhabditis bacteriophora were used as outgroup taxa.
Sequence and phylogenetic analysis
All nucleotide sequences were edited and assembled using Seq-Man II software (DNASTAR, Madison, WI, USA). Subsequently, multiple-sequence alignment with ClustalW and trimmed sequences was performed in MEGA version 7.0 (Kumar et al., Reference Kumar, Stecher and Tamura2016). The 66-kDa protein sequence was blasted in the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm species identification of Angiostrongylus (A. cantonensis and A. malaysiensis).
Phylogenetic trees were constructed via the maximum-likelihood (ML) and Bayesian inference (BI) methods. The ML tree with the Tamura–Nei model (Tamura and Nei, Reference Tamura and Nei1993) was generated via 1000 bootstrap replicates in MEGA version 7.0 (Kumar et al., Reference Kumar, Stecher and Tamura2016). The BI tree was constructed using the MrBayes 3.2.0 program (Ronquist et al., Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2012). The Bayesian posterior probabilities (BPPs) were estimated using Markov chain Monte Carlo analysis, which was run for 10 000 000 generations with data sampling every 500 generations, discarding the first 1000 sampled trees as burn-in (Monte et al., Reference Monte, Simões, Oliveira, Novaes, Thiengo, Silva, Estrela and Maldonado2012). The final phylogenetic trees were viewed and edited in FigTree v.1.4. The nucleotide variation and P distance of A. cantonensis and A. malaysiensis were calculated using the resultant alignment in MEGA version 7.0.
Genetic analysis
The sequences of a 66-kDa protein-encoding gene obtained in the present study together with the sequences downloaded from GenBank were grouped into 3 datasets to analyse the genetic population and the transmission relationships between the host and parasite. Group 1 included all sequences of A. cantonensis (n = 53) and A. malaysiensis (n = 64) populations from different countries. Group 2 consisted of all sequences of A. cantonensis (n = 32) and A. malaysiensis (n = 60) from different regions of Thailand, and group 3 included all currently available sequences of A. cantonensis and A. malaysiensis isolated from intermediate land snail hosts and definitive rodent hosts from different countries.
The genealogical relationships were estimated by using a haplotype network constructed in Network 5.0.1.1 (http://www.fluxus-engineering.com) based on the median-joining algorithm (Bandelt et al., Reference Bandelt, Forster and Rohl1999). Apart from the A. cantonensis and A. malaysiensis 66-kDa protein-encoding sequences in this study, we also included the nucleotide sequences of these worms deposited in GenBank by Eamsobhana et al. (Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). The haplotype nomenclature used in this study was the same as that used by Eamsobhana et al. (Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019).
Genetic diversity indices, e.g. haplotype number, segregating sites, haplotype diversity and nucleotide diversity, were computed and generated by DnaSp version 5 (Librado and Rozas, Reference Librado and Rozas2009) and ARLEQUIN version 3.5.1.2 (Excoffier and Lischer, Reference Excoffier and Lischer2010). Genetic differentiation between Angiostrongylus from different rodent host species was investigated by comparing genetic divergence within rodent host species based on the K2P model in MEGA version 7.0 (Kumar et al., Reference Kumar, Stecher and Tamura2016).
Results
Molecular identification of Angiostrongylus spp.
The molecular identification of Angiostrongylus spp. based on the 66-kDa protein gene was congruent with the morphological identification. PCR-based analysis and sequencing of the 66-kDa protein gene were performed together with a BLASTN search. Fourteen samples (245 bp) of Angiostrongylus (GenBank accession nos. OM280392–OM280405) were identified as A. cantonensis with the highest similarity (100%) with GenBank accession no. MH562093. In addition, 41 sequences of A. malaysiensis in this study (GenBank accession nos. OM280406–OM280446) showed 99–100% identity to A. malaysiensis (GenBank accession nos. MH562059 and MH562113) after a BLASTN search using 245 bp of the 66-kDa protein gene.
Phylogenetic analyses
The phylogenetic trees of A. cantonensis (53 sequences) and A. malaysiensis (64 sequences) were reconstructed using the BI and ML methods. Both methods revealed congruent topologies. Therefore, here we show the BI tree with posterior probabilities and only the bootstrap values from ML analyses. The phylogenetic trees based on 245 bp of the 66-kDa protein gene showed that Angiostrongylus formed a monophyletic clade with a clear separation between A. cantonensis and A. malaysiensis (Fig. 2). The interspecific distances between the A. cantonensis and A. malaysiensis sequences ranged from 0.82 to 2.86%, with a total of 16 variable sites found (Table 1).

Fig. 2. Bayesian tree of A. cantonensis and A. malaysiensis based on 66-kDa protein sequences (245 bp). The BPPs (left) and ML bootstrap values (right) are represented at each node of the phylogenetic tree. Bold letters indicate the sequences obtained in this study. Ancylostoma caninum and Heterorhabditis bacteriophora were used as the outgroup. Ac, A. cantonensis; Am, A. malaysiensis; CPM, Chaiyaphum; CRI, Chiang Rai; KPT, Kamphaeng Phet; PRE, Phrae; TH, Thailand.
Table 1. Haplotypes and their variable nucleotide positions in the 66-kDa protein gene of Angiostrongylus cantonensis and Angiostrongylus malaysiensis

Bold letters indicate the new haplotypes obtained in the present study.
Genetic variation of A. cantonensis and A. malaysiensis populations from different countries
Based on previous studies of A. cantonensis, 13 haplotypes (Ac66-1 to Ac66-13) were classified, and sequences were deposited in GenBank, i.e. haplotypes Ac66-1 to Ac66-4, and Ac66-12 consisted of sequences covering Thailand, China, Japan and the United States, and haplotypes Ac66-8 to Ac66-11 were found only in the United States. In the current haplotype network analyses, our 14 sequences together with 39 sequences from previous studies retrieved from GenBank revealed 14 haplotypes (Ac66-1 to Ac66-14) (Fig. 3). Among the 14 sequence samples obtained in the present study, 9 and 4 sequences belonged to haplotypes Ac66-1 and Ac66-2, respectively. In addition, 1 sequence obtained in the present study was identified as a new haplotype named Ac66-14. A comparison of nucleotide sequences between this new haplotype and 13 previously reported haplotypes is presented in Table 1. The genetic distances among the haplotypes varied from 0 to 0.016 (Table 2). Of these, 10 haplotypes were unique (Ac66-3 to Ac66-9, Ac66-11, Ac66-13 and Ac66-14), and 4 were shared by at least 2 populations (Ac66-1, Ac66-2, Ac66-10 and Ac66-12). The most widely distributed haplotype, Ac66-1, was shared among samples from Thailand, Japan and the United States. Haplotype Ac66-2 was shared between samples from Thailand and Japan. Haplotype Ac66-10 was shared between samples from China and the United States. Haplotype Ac66-12 was shared between samples from Thailand and China. The haplotype diversity in each population ranged from 0.7117 in Thailand to 0.9333 in the United States, with an average of 0.7903. The nucleotide diversity in each population ranged from 0.0035 in China to 0.0076 in the United States, with an average of 0.0054 (Table 3).

Fig. 3. Median-joining haplotype networks of A. cantonensis and A. malaysiensis from Thailand and other geographical regions inferred from 66-kDa protein sequences. Each haplotype is represented by a circle, and circle sizes are proportional to haplotype frequency. Colours indicate the geographic origin of the haplotypes. Each mutation between haplotypes is represented by a bar. Median vectors (small red dots) represent ancestral haplotypes that are either not sampled or missing haplotypes.
Table 2. Genetic distance among haplotypes of A. cantonensis based on 66-kDa protein sequences

Table 3. Genetic diversity of A. cantonensis and A. malaysiensis from Thailand and other geographical regions based on 66-kDa protein sequences

Thirteen haplotypes (Am66-1 to Am66-13) of A. malaysiensis were identified from 41 sequences obtained in the present study together with 23 sequences from GenBank. Among the 41 samples from the present study, 10 sequences belonged to haplotype Am66-1, and 31 sequences belonged to the 5 new haplotypes, including Am66-9 (19 sequences), Am66-10 (9 sequences), Am66-11 (1 sequence), Am66-12 (1 sequence) and Am66-13 (1 sequence). A comparison of nucleotide sequences between the 5 new haplotypes identified in the present study and 8 previously reported haplotypes is presented in Table 1. The genetic distances between the haplotypes varied from 0 to 0.016 (Table 4). Of these, 12 haplotypes were unique (Am66-2 to Am66-13), and 1 (Am66-1) was shared between samples from Malaysia and Thailand (Fig. 3). The haplotype diversity in each population ranged from 0 in Malaysia to 0.8096 in Thailand, with an average of 0.7981. The nucleotide diversity in each population ranged from 0 in Malaysia to 0.0056 in Thailand, with an average of 0.0054 (Table 3).
Table 4. Genetic distance among haplotypes of A. malaysiensis based on 66-kDa protein sequences

Genetic variation of A. cantonensis and A. malaysiensis isolated from different regions of Thailand
The analysis of 32 A. cantonensis sequences from Thailand identified 6 haplotypes (Ac66-1 to Ac66-4, Ac66-12 and Ac66-14). Among the A. cantonensis haplotypes in Thailand, Ac66-1 was the most common and was widely distributed in 4 regions (central, north, northeast and south). Another common haplotype was Ac66-2, which was found in several geographical localities in 3 regions (central, northeast and south), and haplotype Ac66-4 was found in the northeast and south regions of Thailand. The remaining haplotypes were found to be unique in a particular isolate, such as haplotype Ac66-3, which was found only in the central isolate, Ac66-12, which was unique to the western isolate, and Ac66-14, which was found only in the northeast isolate (Fig. 4). The haplotype diversity in each population ranged from 0 in the population from the north and west regions to 0.7333 in the population from the south, with an average of 0.6613. The nucleotide diversity in each population ranged from 0 in the north and west regions to 0.8205 in the northeast region, with an average of 0.0034 (Table 5).

Fig. 4. Median-joining haplotype networks of A. cantonensis and A. malaysiensis from different regions of Thailand inferred from 66-kDa protein sequences. Each haplotype is represented by a circle, and circle sizes are proportional to haplotype frequency. Colours indicate the geographic origin of the haplotypes. Each mutation between haplotypes is represented by a bar. Median vectors (small red dots) represent ancestral haplotypes that are either not sampled or missing haplotypes.
Table 5. Genetic diversity of A. cantonensis and A. malaysiensis from different regions of Thailand based on 66-kDa protein sequences

Thirteen haplotypes (Am66-1 to Am66-13) of A. malaysiensis were identified from 60 Thailand sequences. Among the A. malaysiensis haplotypes, Am66-4 was the most common and was widely distributed in 4 regions (north, northeast, south and west). Another common haplotype was Am66-1, which was found in several geographical localities in 3 regions (central, north and west), and haplotype Am66-6 was found in isolates from the central and south regions of Thailand. The remaining haplotypes were found to be unique in a particular isolate, such as haplotypes Am66-2, Am66-3 and Am66-8, which were found only in the northeast isolate; Am66-5 and Am66-7, which were unique to the south isolate and Am66-9 to Am66-13, which were present only in the north isolate (Fig. 4). The haplotype diversity in each population ranged from 0.6667 in the population from the west region to 1.0000 in the population from the central and northeast regions, with an average of 0.8096. The nucleotide diversity in each population ranged from 0.0027 in the west region to 0.0082 in the central and northeast regions, with an average of 0.0056 (Table 5).
Genetic variation of A. cantonensis and A. malaysiensis isolated from snail intermediate and rodent definitive hosts
This group consisted of 53 and 64 sequences that were isolated from intermediate land snail hosts and definitive rodent hosts, respectively. In the land snail A. fulica, 12 A. cantonensis and 41 A. malaysiensis sequences were classified into 3 haplotypes (Ac66-1, Ac66-2 and Ac66-14) and 6 haplotypes (Am66-1, Am66-9 to Am66-13), respectively (Fig. 5). Among the A. cantonensis haplotypes, 3 haplotypes (Ac66-1, Ac66-2 and Ac66-14) were found in A. fulica in Chaiyaphum province of Thailand. Of these, only 2 haplotypes (Ac66-1 and Ac66-2) were found in rodent hosts from different countries, such as haplotype Ac66-1 found in isolates from Thailand, Japan and the United States, and Ac66-2 was present in isolates from Thailand and Japan. In A. malaysiensis, Am66-1 was the most widely distributed in A. fulica in 2 provinces (Chiang Rai and Phrae) in northern Thailand. In addition, this haplotype was found in rodent hosts from the central province (Bangkok), 2 northern provinces (Chiang Rai and Mae Hong Son) and 2 western provinces (Kanchanaburi and Tak) of Thailand and in Pahang in Malaysia.

Fig. 5. Median-joining haplotype networks of A. cantonensis and A. malaysiensis from different land snail and rodent host species inferred from 66-kDa protein sequences. Each haplotype is represented by a circle, and circle sizes are proportional to haplotype frequency. Colours indicate the geographic origin of the haplotypes. Each mutation between haplotypes is represented by a bar. Median vectors (small red dots) represent ancestral haplotypes that are either not sampled or missing haplotypes.
For the rodent host species, the haplotype diversity of A. cantonensis ranged from 0 in Bandicota sp. to 0.8889 in Rattus species. Nucleotide diversity ranged from 0 in Bandicota sp. to 0.0071 in Rattus norvegicus. Among the 13 haplotypes identified, 8 were unique, and 5 haplotypes were shared by at least 2 rodent host species. Rattus norvegicus possessed the highest number (5 haplotypes) of unique haplotypes (Fig. 5, Table 6). Genetic divergence within the rodent host species based on the K2P model ranged from 0 to 1.66%, with a mean of 0.47% (Table 6). The greatest within-rodent host genetic divergence (1.66%) was found in R. norvegicus. In A. malaysiensis, the haplotype diversity ranged from 0 in Bandicota indica and Rattus tiomanicus to 1.0000 in Bandicota bengalensis, Rattus exulans, Rattus losea and Rattus norvegicus. Nucleotide diversity ranged from 0 in B. indica and R. tiomanicus to 0.0082 in R. exulans. Among the 8 haplotypes identified, 5 were unique, and 3 haplotypes were shared by at least 2 rodent host species. Rattus norvegicus possessed the highest number (3 haplotypes) of unique haplotypes (Fig. 5, Table 6). Genetic divergence within the rodent host species based on the K2P model ranged from 0 to 1.24%, with a mean of 0.41% (Table 6). The greatest within-rodent host genetic divergence (1.24%) was found in R. rattus.
Table 6. Haplotype diversity (h), nucleotide diversity (h) and genetic divergence based on 66-kDa sequences between A. cantonensis and A. malaysiensis from different host species

NA, not analysed.
Discussion
The advantage of the 66-kDa protein gene as a genetic marker to determine genetic diversity and phylogeny between and within Angiostrongylus populations (A. cantonensis, A. malaysiensis and A. costaricensis) was previously noted (Eamsobhana et al., Reference Eamsobhana, Lim, Zhang, Gan and Yong2010, Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). In this study, identification of Angiostrongylus based on a partial sequence of a 66-kDa protein-encoding gene was confirmed by 99–100% sequence identity after BLASTN searches. Angiostrongylus cantonensis and A. malaysiensis are closely related in terms of morphological and genetic characteristics and also share similarities in their life cycles (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020; Watthanakulpanich et al., Reference Watthanakulpanich, Jakkul, Chanapromma, Ketboonlue, Dekumyoy, Lv, Chan, Thaenkham and Chaisiri2021). Both species utilize the same definitive and intermediate host species (Bhaibulaya and Techasoponmani, Reference Bhaibulaya and Techasoponmani1972). In addition, mixed infections with both A. cantonensis and A. malaysiensis have been widely recorded in snail intermediate hosts and rodent definitive hosts (Jakkul et al., Reference Jakkul, Chaisiri, Saralamba, Limpanont, Dusitsittipon, Charoennitiwat, Chan and Thaenkham2021; Watthanakulpanich et al., Reference Watthanakulpanich, Jakkul, Chanapromma, Ketboonlue, Dekumyoy, Lv, Chan, Thaenkham and Chaisiri2021). The 66-kDa protein gene is undoubtedly suitable for discrimination between A. cantonensis and A. malaysiensis because the phylogenetic trees clearly place these 2 species into separate clades. Our findings are similar to those of previous reports (Eamsobhana et al., Reference Eamsobhana, Lim, Zhang, Gan and Yong2010, Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). This suggests the advantage of the 66-kDa protein sequence for the identification of A. cantonensis and A. malaysiensis. Interestingly, the use of the 66-kDa protein gene as the genetic marker was successful in discriminating the third-stage larvae of A. cantonensis and A. malaysiensis.
The level of genetic divergence of the 66-kDa protein sequences between A. cantonensis and A. malaysiensis ranged from 0.82 to 2.86%, which was relatively low when compared with other genetic markers. Our findings are similar to those of Eamsobhana et al. (Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019), who reported that the genetic distance between A. cantonensis and A. malaysiensis using 66-kDa protein sequences was approximately 3.27% (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). A lower genetic divergence (0–1.0%) in the nuclear 18S rRNA gene was also noted between A. cantonensis and A. malaysiensis (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020). In contrast, the genetic divergence between A. cantonensis and A. malaysiensis was relatively high based on COI (9.8–16.4%), cytb (10.9–12.2%), 12S rRNA (6.8–7.9%), 16S rRNA (7.9–10.0%) and ITS2 (15.1–15.7%) sequences (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020). However, several reports have shown that the nuclear 18S rRNA gene can be used to discriminate between A. cantonensis and A. malaysiensis, which are clearly distinguished into their clades using this marker despite the low interspecies genetic distance (Fontanilla and Wade, Reference Fontanilla and Wade2008; Tokiwa et al., Reference Tokiwa, Harunari, Tanikawa, Komatsu, Koizumi, Tung, Suzuki, Kadosaka, Takada, Kumagi, Akao and Ohta2012; Rodpai et al., Reference Rodpai, Intapan, Thanchomnang, Sanpool, Sadaow, Laymanivong, Aung, Phosuk, Lammaunwai and Maleewong2016), which is comparable to the results we obtained using the 66-kDa protein gene.
Previous studies of third-stage larvae of A. cantonensis isolated from A. fulica snails in 8 provinces of Thailand using 8 random-amplified polymorphic DNA-PCR markers revealed high levels of genetic diversity and low levels of gene flow in A. cantonensis populations (Thaenkham et al., Reference Thaenkham, Pakdee, Nuamtanong, Maipanich, Pubampen, Sanguankiat and Komalamisra2012). The worms from these 8 localities were divided into 2 groups with statistically significant genetic differentiation of the 2 populations: group 1 contained A. cantonensis from Chanthaburi, Chiang Mai, Khon Kaen, Narathiwat Nong Khai and Prachuap Khiri Khan provinces, and group 2 contained A. cantonensis from Kanchanaburi and Lop Buri provinces. Similarly, Vitta et al. (Reference Vitta, Srisongcram, Thiproaj, Wongma, Polsut, Fukruksa, Yimthin, Mangkit, Thanwisai and Dekumyoy2016) reported third-stage larvae of A. cantonensis from freshwater and land snails in 19 distinct geographical areas of Thailand using COI sequences, revealing 2 different origins of A. cantonensis in Thailand: group 1 contained A. cantonensis from Kamphaeng Phet, Phetchabun, Tak and Thailand ac4, and group 2 contained A. cantonensis from Kalasin, Kamphaeng Phet, Phitsanulok, Tak and AC Thai. Nonetheless, haplotypes of groups 1 and 2 were found in the same areas (Kamphaeng Phet and Tak provinces). The results indicate the occurrence of restricted gene flow between localities.
A recent study reported the presence of 13 distinct 66-kDa protein sequence haplotypes (Ac66-1 to Ac66-13) from A. cantonensis in several parts of the world (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). Haplotype Ac66-1 was the most common haplotype in A. cantonensis from Thailand, Japan and the United States. Haplotype Ac66-5 was reported in Japan, while haplotypes Ac66-8, Ac66-9 and Ac66-11 were reported in the United States. Haplotype Ac66-10 was reported in China and the United States, and haplotype Ac66-13 was found only in China. In Thailand, A. cantonensis did not cluster unequivocally according to their geographical origin, as 7 haplotypes (Ac66-1 to Ac66-4, Ac66-6, Ac66-7 and Ac66-12) from 10 geographical regions of Thailand were found. The A. cantonensis haplotypes from Bangkok and Phitsanulok provinces in the central region, Surat Thani province in the south, and the Thailand laboratory strain (originating from Khon Kaen in the northeast) were variable. Moreover, 4 haplotypes were found confined to a single locality: Ac66-3 in Phitsanulok province (central region); Ac66-6 and Ac66-7 in the Thailand laboratory strain and Ac66-12 in Prachuap Khiri Khan province (west region) (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). In the present study, haplotype Ac66-1 was found in Kamphaeng Phet province (2 specimens) in the central region and Chaiyaphum province (7 specimens) in the northeast region of Thailand. Additionally, haplotype Ac66-1 was previously reported in 3 central provinces (Bangkok, Lop Buri and Phitsanulok), 2 southern provinces (Ranong and Surat Thani) and in the northern province (Chiang Mai) (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). This Ac66-1 haplotype was dominant in Thailand. The other 4 specimens (haplotype Ac66-2) from Chaiyaphum province in the present study were also reported from 2 central provinces (Bangkok and Samut Prakan) and the south province (Surat Thani). Importantly, 1 new haplotype (Ac66-14) was identified in 1 specimen from Chaiyaphum province of Thailand. Incorporation of the genetic data of the 66-kDa protein gene from A. cantonensis obtained in the present and previous studies (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019) revealed sharing of dominant haplotypes (Ac66-1 and Ac66-2), suggesting a common origin.
Based on the 66-kDa protein sequence from A. malaysiensis, previous studies reported 8 haplotypes (Am66-1 to Am66-8) in Thailand and Malaysia (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). In this study, 13 haplotypes were identified, with 5 haplotypes being new. One haplotype (Am66-1) of A. malaysiensis from Phrae (3 specimens) and Chiang Rai (7 specimens) provinces in the north region was previously reported from Thailand and Malaysia (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). Five new haplotypes (Am66-9, Am66-10, Am66-11, Am66-12 and Am66-13) were reported from 31 specimens in Chiang Rai province of Thailand. The most common haplotype detected in Thailand and Malaysia was Am66-1. In Thailand, 66-kDa protein haplotypes (Am66-1 to Am66-8) were distributed at random throughout the country, and Am66-1 was the most widely distributed in 2 northern provinces (Chiang Rai and Mae Hong Son), 2 western provinces (Kanchanaburi and Tak) and in a central province (Bangkok). In addition, the A. malaysiensis haplotypes from Bangkok in central Thailand, Mae Hong Son in north Thailand, Nong Khai in northeast Thailand, Satun in south Thailand and Tak in west Thailand showed variable 66-kDa haplotype diversity. Moreover, 5 haplotypes were found confined to a single locality: Am66-2, Am66-3 and Am66-8 in Nong Khai province (northeast region); Am66-5 in Phang Nga province (south region) and Am66-7 in Satun province (south region) (Eamsobhana et al., Reference Eamsobhana, Yong, Song, Gan, Prasartvit and Tungtrongchitr2019). Therefore, it was difficult to conclude the relationship between the haplotype of A. malaysiensis and geographic areas in Thailand. A larger sample size of the 66-kDa protein-encoding gene sequence may reveal a clearer relationship between haplotypes and localization in Thailand.
High haplotype diversity in a 66-kDa protein gene for A. cantonensis (14 haplotypes) and A. malaysiensis (13 haplotypes) has also been observed in other genetic markers. Twenty cytb haplotypes have been reported for A. cantonensis from several parts of the world (Dusitsittipon et al., Reference Dusitsittipon, Thaenkham, Watthanakulpanich, Adisakwattana and Komalamisra2015; Yong et al., Reference Yong, Eamsobhana, Song, Prasartvit and Lim2015; Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019). For the COI gene, 16 haplotypes were observed in A. cantonensis globally (Eamsobhana et al., Reference Eamsobhana, Song, Yong, Prasartvit, Boonyong and Tangtrongchitr2017), and 9 haplotypes were observed in A. malaysiensis from Laos, Malaysia, Myanmar and Thailand (Rodpai et al., Reference Rodpai, Intapan, Thanchomnang, Sanpool, Sadaow, Laymanivong, Aung, Phosuk, Lammaunwai and Maleewong2016; Eamsobhana et al., Reference Eamsobhana, Yong, Song, Prasartvit, Boonyong and Tungtrongchitr2018). For the 12S rRNA gene of Angiostrongylus in Thailand, the average genetic variation was 0.5% with 6 haplotypes in a population of A. cantonensis (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020). Similarly, using the 16S rRNA gene, an average genetic variation of 2.2% with 6 haplotypes within 1 population of A. cantonensis and an average genetic variation of 1.7% with 6 haplotypes within 4 populations of A. malaysiensis were found (Chan et al., Reference Chan, Chaisiri, Dusitsittipon, Jakkul, Charoennitiwat, Komalamisra and Thaenkham2020). Comparatively, we found that the 66-kDa protein gene resulted in 3 haplotypes within 2 populations of A. cantonensis (Chaiyaphum and Kamphaeng Phet provinces). Moreover, 6 haplotypes were found in 2 populations of A. malaysiensis (Phrae and Chiang Rai provinces). Higher intraspecific genetic variation levels and more haplotypes might be observed if more A. cantonensis and A. malaysiensis specimens are sampled from other localities.
To investigate the distribution of haplotypes in hosts, median-joining networks were constructed to analyse all currently available 66-kDa protein sequences of A. cantonensis and A. malaysiensis isolated from snail intermediate hosts (A. fulica) and rodent definitive hosts from different countries. In A. cantonensis, 2 haplotypes (Ac66-1 and Ac66-2) were shared between A. cantonensis isolated from A. fulica (from Chaiyaphum province of Thailand) and rodent hosts (from Thailand, Japan and the United States). In A. malaysiensis, Am66-1 was the most widely distributed haplotype in A. fulica in 2 northern provinces (Chiang Rai and Phrae) of Thailand. In addition, this haplotype was found in rodent hosts from the central province (Bangkok), 2 northern provinces (Chiang Rai and Mae Hong Son) and 2 western provinces (Kanchanaburi and Tak) of Thailand and in Pahang in Malaysia. This could be considered the result of range expansion of genomic origin. These findings indicate that lineage-specific A. cantonensis and A. malaysiensis have been spreading across Thailand. The transmission of this nematode has been linked to the dispersal of invasive hosts (Tokiwa et al., Reference Tokiwa, Harunari, Tanikawa, Komatsu, Koizumi, Tung, Suzuki, Kadosaka, Takada, Kumagi, Akao and Ohta2012). The increased presence of A. cantonensis in the country is likely a result of the rapid spread of its intermediate host, A. fulica, contributing to the dispersion of this parasite and infection of the definitive host (Thiengo et al., Reference Thiengo, Maldonado, Mota, Torres, Caldeira, Carvalho, Oliveira, Simões, Fernandez and Lanfredi2010; Dumidae et al., Reference Dumidae, Janthu, Subkrasae, Dekumyoy, Thanwisai and Vitta2019, Reference Dumidae, Subkrasae, Ardpairin, Thanwisai and Vitta2021). Invasion by A. fulica facilitates the establishment of the life cycle of the parasite and thus increases the chances for exposure of native snails to A. cantonensis in existing endemic areas. In addition, this invasive snail accelerates the spread of A. cantonensis to new areas since it rapidly expands the parasite's range (Lv et al., Reference Lv, Zhang, Liu, Zhang, Steinmann, Zhou and Utzinger2009). This phenomenon is described as one of the primary causes of the spread of oeosinophilic meningitis (Maldonado et al., Reference Maldonado, Simões, Oliveira, Motta, Fernandez, Pereira, Monteiro, Torres and Thiengo2010). Invasive snails and rodents are implicated in an increase in the distribution of A. cantonensis in Brazil (Thiengo et al., Reference Thiengo, Maldonado, Mota, Torres, Caldeira, Carvalho, Oliveira, Simões, Fernandez and Lanfredi2010), Spain (Foronda et al., Reference Foronda, López-González, Miquel, Torres, Segovia, Abreu-Acosta, Casanova, Valladares, Mas-Coma, Bargues and Feliu2010), China (Yang et al., Reference Yang, Wu and Lun2013), Uganda (Mugisha et al., Reference Mugisha, Bwangamoi and Cranfield2012), Japan (Tokiwa et al., Reference Tokiwa, Hashimoto, Yabe, Komatsu, Akao and Ohta2013) and the United States (York et al., Reference York, Creecy, Lord and Caire2015). The hypothesis that Angiostrongylus has achieved global-scale dispersal with various organisms/hosts or vectors is largely influenced by human transportation (Monte et al., Reference Monte, Simões, Oliveira, Novaes, Thiengo, Silva, Estrela and Maldonado2012; Tokiwa et al., Reference Tokiwa, Harunari, Tanikawa, Komatsu, Koizumi, Tung, Suzuki, Kadosaka, Takada, Kumagi, Akao and Ohta2012; Dusitsittipon et al., Reference Dusitsittipon, Thaenkham, Watthanakulpanich, Adisakwattana and Komalamisra2015).
Comparisons of genetic divergence among rodent host species found unique and shared haplotypes in different rodent species. All rodent host species included in the present study shared at least 1 common 66-kDa protein haplotype. Our findings showed that 8 haplotypes were unique and 5 haplotypes were shared in A. cantonensis, whereas 5 haplotypes of A. malaysiensis were found to be unique, and 3 haplotypes were shared by at least 2 rodent host species. The high degree of haplotype uniqueness suggests that there are some limitations to the spread of genomic origin among rodent host species. Many rodent species serve as definitive hosts for A. cantonensis and A. malaysiensis and are capable of highly promoting the distribution and intraspecific transfer of this parasite (Eamsobhana et al., Reference Eamsobhana, Yong, Prasartvit, Wanachiwanawin and Boonyong2016). The limited dispersal of rodent hosts might be expected to limit the genomic origin of their worm parasites, resulting in genetic structure over small geographical scales (Pocock et al., Reference Pocock, Hauffe and Searle2005; Gardner-Santana et al., Reference Gardner-Santana, Norris, Fornadel, Hinson, Klein and Glass2009). However, data on the genetic diversity of Angiostrongylus remain scarce in invaded areas (Simões et al., Reference Simões, Monteiro, Sanchez, Thiengo, Garcia, Costa-Neto, Luque and Maldonado2011; Monte et al., Reference Monte, Simões, Oliveira, Novaes, Thiengo, Silva, Estrela and Maldonado2012; Moreira et al., Reference Moreira, Giese, Melo, Simões, Thiengo, Maldonado and Santos2013; Dalton et al., Reference Dalton, Fenton, Cleveland, Elsmo and Yabsley2017). The pathogenicity of A. cantonensis against laboratory hosts varies between different A. cantonensis genetic strains (Lee et al., Reference Lee, Chung, Wang, Lin, Wang, Tu, Wu and Yen2014). Whether this dominant haplotype presents any difference in pathogenicity compared to other haplotypes remains to be clarified. Further studies are needed to clarify whether this haplotype has a different pathogenicity that may contribute to its evolution.
Conclusion
This study further confirmed the presence of 66-kDa protein genetic diversity in various geographical isolates of A. cantonensis and A. malaysiensis. We demonstrate the utility of the 66-kDa protein-encoding gene as a genetic marker for species discrimination in the larval stage, as it clearly discriminated A. cantonensis and A. malaysiensis into separate clades. Importantly, we identified 1 and 5 new haplotypes from A. cantonensis and A. malaysiensis, respectively. Our findings revealed that the 66-kDa protein gene has sufficient intraspecific genetic variation to be considered a genetic marker for future Angiostrongylus population-level studies.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182022001573
Data
All relevant data are within the paper and its supporting information file.
Acknowledgement
We acknowledge the Department of Microbiology and Parasitology, Faculty of Medical Science, Naresuan University for supporting facilities.
Author's contributions
A. D. and A. V. conceived and designed the study. A. D., C. S., J. A., A. T. and A. V. performed the experiments. A. D., A. T. and A. V. analysed the data. A. T. and A. V. provided materials. A. D. and A. V. wrote the paper.
Financial support
A part of this study was funded by Naresuan University (grant number R2562B079).
Conflict of interest
We have no conflict of interest to declare.
Ethical standards
The experimental protocol for the use of animals in this study was approved by the Center for Animal Research of Naresuan University (Project Ethics Approval No. NU-AE650402).













 Open access
Open access