


I Introduction
Health care’s digital transformation – accelerated, but by no means initiated, by the COVID-19 pandemic – has garnered attention as patients increasingly expect remote care options. A preponderance of digital health applications and connected sensors are poised to transform how health care is delivered in contexts outside of the hospital or clinic.Footnote 1
The digitization of health care delivery and medical technology raises questions about the safety of digital medical devices and how regulators monitor and respond to safety questions. One concern is that introducing software components to previously analog medical devices may create unexpected complexity or harm. For example, patients have died due to drug overdoses caused by “key bounce” in infusion pump software, whereby software incorrectly interprets a single keystroke as multiple keystrokes, resulting in patients receiving far more medicine than intended.Footnote 2
Even given the known safety concerns associated with digital products, the existing infrastructure for tracking medical device safety may not be well equipped to monitor the safety of products that are (increasingly) used outside of traditional health care facilities. Most post-market surveillance – that is, ongoing regulatory oversight beyond initial regulatory approval/clearance – in the United States takes the form of adverse event reporting by device manufacturers and (health care) user facilities or post-approval trials conducted by manufacturers.Footnote 3 Given that post-market surveillance primarily relies on the vigilance of manufacturers and health care providers, regulators may miss important safety signals as medical technologies are moved from health care facilities to patients’ homes.
These safety challenges have important implications for remote patient monitoring (RPM) tools. RPM is the collection of physiological measures that can be shared with health care providers – both actively by patients (e.g., by taking measurements and entering data at home) or passively with connected devices (which may automatically enter such data into a relevant database).Footnote 4 RPM encompasses the use of both combined hardware–software products, such as connected sensors, as well as standalone software tools.
Here, we focus specifically on the subset of RPM and other software-driven products that meet the definition of a medical device in the United States and, therefore, are subject to regulation by the US Food and Drug Administration (FDA). By focusing on regulated diagnostic and therapeutic devices, we specifically focus on products used in patients’ formal health care delivery instead of more consumer-health/wellness-oriented digital products. In other words, this chapter does not consider the overwhelmingly large set of consumer health apps that may or may not be verified or validated.Footnote 5 Importantly, we consider all medical devices containing software – both standalone software tools (often called “software as medical devices,” or SaMDs) as well as combination hardware–software products (“software in medical devices,” or SiMDs). In doing so, we follow the definition of “software-driven medical devices” (SdMDs) introduced by Gordon and Stern (2019) (which includes both SaMDs and SiMDs) and consider all SdMDs subject to FDA oversight.Footnote 6 Relative to digital diagnostics and therapeutics used outside of traditional clinical settings, our sample represents a highly relevant set of products, but is almost certainly a “super-set” of those regulated devices used in remote diagnosis and care.
The chapter proceeds as follows. First, we provide a brief overview of post-market surveillance of regulated medical devices in the United States and present data on post-market outcomes from recent years. Next, in detailed regulatory data, we identify SdMDs among regulated devices and document trends in their approvals, as well as the associated post-market safety issues. Finally, we conclude with a discussion of the implications of our findings for regulatory policy and the future of post-market surveillance for SdMDs.
II Post-Market Surveillance Activities and Regulatory Data
For regulated medical technologies, post-market surveillance plays an important role in ensuring that products continue to be safe and effective. The FDA’s Center for Devices and Radiological Health (CDRH) notes that post-market surveillance activities may include “tracking systems, reporting of device malfunctions, serious injuries or deaths, and registering the establishments where devices are produced or distributed.” Further, post-market requirements may also include surveillance studies and additional post-approval studies that were deemed to be required at the time of device approval.Footnote 7 We briefly summarize these activities and the types of publicly available data that they generate before turning to an empirical analysis.
Under 21 USC § 360I, the FDA has the authority to require manufacturers to engage in various post-market activities. These may be required at either the time of approval/clearance of a new device or sometime thereafter. An FDA Guidance Document further outlines best practices for the medical device industry with respect to several aspects of post-market surveillance,Footnote 8 including surveillance planning, interim reporting, and the implications of failing to comply with post-market reporting requirements. The following sections provide an overview of the various post-market activities that the FDA may require.
A Post-Market Trials and Registries
Two common ways in which manufacturers and regulators continue to monitor the ongoing safety and effectiveness of medical devices are via post-market clinical trials and patient registries.
One or more post-approval studies may be required by regulators at the time of a Pre-Market Approval (PMA), Humanitarian Device Exemption (HDE), or Product Development Protocol (PDP) application. The FDA may require that manufacturers commit to conducting such studies before it grants regulatory approval, and failure to complete studies may be grounds for the FDA to withdraw a device’s approval.Footnote 9 For example, the Post-Approval Study on Patients Who Received a HeartWare HVAD® During IDE Trials (HW-PAS-03), a multi-center study sponsored by the device’s manufacturer, provided continued evaluation and follow-up on patients who had received the HeartWare® Ventricular Assist System during earlier clinical trials.Footnote 10 The FDA may request that post-approval studies be conducted for both moderate- and high-risk devices. In practice, post-market studies are often delayed or terminated after the manufacturer changes the indication for use of the studied medical device.Footnote 11
Patient registries may be device-specific or embedded in larger surveillance initiatives. For example, as a condition for the approval of transcatheter heart valves, the FDA required all manufacturers to “continue to follow patients enrolled in their randomized studies for 10 years to further monitor transcatheter aortic valve safety and effectiveness….” As part of this initiative, the manufacturers agreed to participate in the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy (TVT) Registry.Footnote 12
B Plant Inspections
Another important component of post-market medical device regulation includes the inspection of plants where devices with hardware components are manufactured. Ball et al. (2017) summarized the rationale for manufacturing plant inspections by noting that “governments cannot feasibly sample every manufactured product before its release to customers; therefore, they frequently depend on plant inspections to appraise a plant’s quality systems.”Footnote 13
Generally speaking, device-manufacturing plant inspections are conducted according to the process described in the Quality System Inspection Technique Guide, which, in turn, follows the requirements contained within 21 CFR § 820.Footnote 14 Such plant inspections involve the detailed documentation of various processes – including those associated with quality system requirements, various forms of controls (e.g., design, production, and process), corrective and preventative actions, and so on. Notably, investigators do not inspect actual products, but, instead, examine the systems that guide the device manufacturing process.
Inspectors typically arrive at a plant unannounced, tour the facility, interview managers, and perform a process documentation review. There are three different types of such inspections: (1) Surveillance inspections – those that occur regularly and routinely to assess plant quality; (2) compliance inspections – those that are part of the establishment of new or modified manufacturing processes or new product launches; and (3) complaint inspections – those that occur in response to serious complaints by customers/device users.Footnote 15 In response to inspections, remedial actions may or may not be indicated; remedial actions may be “voluntary” or “official,” depending on the severity of issues identified.Footnote 16
C Medical Device Reporting
Once devices are legally marketed, a system of voluntary and mandatory medical device reporting serves to track adverse events and identify emergent safety issues. The FDA receives several hundred thousand medical device reports (MDRs) related to suspected device-associated malfunctions, injuries, and deaths annually.Footnote 17 These reports are collected in the Manufacturer and User Facility Device Experience (MAUDE) database, which is maintained by the FDA. Reports are mandatory for certain users – namely device manufacturers, importers, and health care facilities – and voluntary for others, including patients, consumers, and clinicians.
MDRs are input into the MAUDE database along with detailed product information, which includes a device’s manufacturer, product code, and FDA clearance/approval identifiers. This information allows individual MDRs to be linked to specific products. Although MDRs and the accompanying MAUDE database represent rich and well-organized sources of information, the FDA warns that the surveillance system may be incomplete, unverified, or inaccurate because of biased reporting, reporting lags, and other factors, and therefore cautions against using MAUDE data to understand the frequency or causality of adverse events. Nevertheless, MAUDE remains an important source of information about product quality issues, and its open-source format lends itself to empirical research in medicine and health policy.Footnote 18
D Recalls
Finally, post-market regulation includes the oversight of formal medical device recalls. Although recalls are typically manufacturer-initiated, they are overseen by the FDA, which classifies recalls according to risk/severity:
Class I recalls (most severe) occur where “there is a reasonable chance that a product will cause serious health problems or death” – for example, a faulty pacemaker lead that would prevent proper functioning.
Class II recalls (moderate severity) occur where “a product may cause a temporary or reversible health problem or where there is a slight chance that it will cause serious health problems or death” – for example, an insufficiently tight surgical clamp.
Class III recalls (low severity) occur where “a product is not likely to cause any health problem or injury” but where an issue nevertheless should be corrected – for example, a labeling issue.Footnote 19
The FDA’s medical device recall database publishes data on all classes of product recalls. The database links recall information to specific clearance/approval decision identifiers, enabling researchers to link a recall to at least one specific previously regulated product.
III Methods for Data Collection and Analysis
In this section we describe the datasets we used to quantify the likelihood of post-market safety events associated with SdMDs and other devices over recent years.
A Data Sources and Sample Construction
We identified all 510(k)-track and PMA-track medical devices (i.e., moderate and high-risk devices) cleared or approved by the FDA from 2008–2018 in the five common regulatory medical specialties (associated with CDRH Advisory Committees of the same name) most likely to include RPM devices: Cardiology, clinical chemistry, gastroenterology, general hospital, and general and plastic surgery. We then identified all recalls and adverse events associated with these devices that occurred between 2008 and 2020 using the FDA’s MAUDE and recall databases, respectively. We limited data from MAUDE to only include adverse events from mandatory reporters to reduce non-random differences in reporting across device types.
B Identifying Software-Driven Medical Devices
We employed a supervised document classification algorithm to identify SdMDs. For each medical device in our sample, we downloaded its associated public statement or summary document from the FDA’s website. These documents are required for all submissions and each “includes a description of the device such as might be found in the labeling or promotional material for the device.”Footnote 20 We then used optical character recognition software to search each document for the word “software” to identify devices with a software component.
This text search technique was demonstrated to work well in manual review: In comparison to a manually coded random sample of summary documents, the document classification had a 0 percent false negative rate, meaning devices flagged as including a software component via supervised document classification always included a software component. Accordingly, we identified a medical device as including a software component if “software” appeared at least once in its public summary of evidence. Additional details on the supervised document classification are provided elsewhere.Footnote 21
C Outcomes of Interest
We focused on two primary outcomes of interest: (1) Class I/II recalls (i.e., those of moderate or greater severity) and (2) mandatorily reported adverse events. For recalls, we identified all class I/II recalls that occurred within two years of regulatory approval/clearance for each device. We chose to use two years of follow-up, as most medical device recalls occur shortly after a medical device comes to market.Footnote 22 For adverse events, we similarly created a count of all adverse events from mandatory reporters in the two years following a device’s clearance/approval.
D Statistical Analysis
We compared differences in adverse events and recalls by software status by performing two-sided, two-sample t-tests comparing the outcomes between SdMDs vs. non-SdMDs. To understand the changes over time, we plotted the number of recalls or adverse events in a given calendar year divided by the number of approvals/clearances in the two preceding years, such that the frequency of outcomes was scaled by the number of devices recently placed on the market in each year. All statistical analyses were performed using data from the entire sample, as well as within individual medical specialties.
IV Results
Our sample included 13,186 medical devices, or 39.46 percent of all medical devices approved or cleared by the FDA during the sample period. During this time, software became increasingly prevalent in medical devices: While we observed variation over time in the total number and share of new SdMDs cleared/approved, all five medical specialties had a greater number and proportion of cleared/approved devices that included a software component in 2020 vs. 2010 (Figure 9.1). For example, 25.7 percent of the cardiovascular devices cleared or approved in 2010 included a software component, vs. 27.8 percent in 2020.
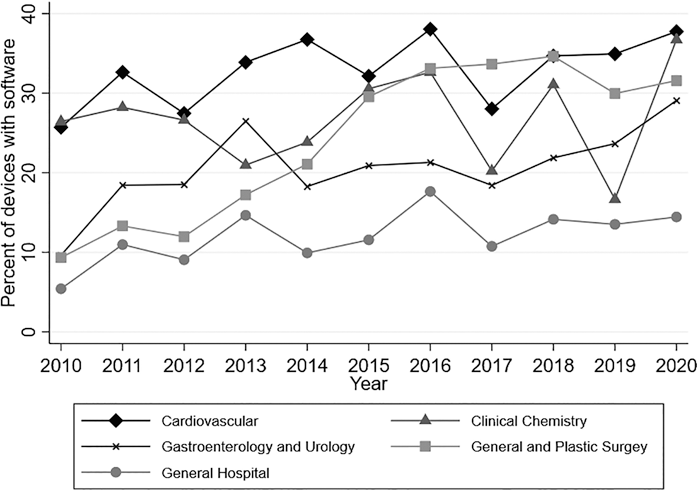
Figure 9.1 Proportion of devices with software by specialty over time
Note: Authors’ analysis of FDA approval and clearance data from 2010–2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of devices approved/cleared).
SdMDs in our sample experienced more adverse events (Figure 9.2) and class I/II recalls (Figure 9.3) than devices without software. The average SdMD had 14.516 associated adverse events from mandatory reporters in the MAUDE database (in its first two years on the market), while the average device without software had 3.524 associated adverse events reported (p = 0.010) (Table 9.1). Similarly, 8.1 percent of SdMDs experienced at least one class I/II recall in the two years following regulatory approval/clearance, vs. 3.6 percent of devices without software (p < 0.001) (Table 9.1).

A: No software.

B: Software.
Note: Authors’ analysis of FDA approval and clearance data and the FDA’s MAUDE database from 2010–2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each year–specialty observation, the total adverse events from mandatory reporters were calculated and then divided by the number of approvals and clearances within that specialty in the preceding two years.
Figure 9.2 Two-year adverse event rates by specialty over time.
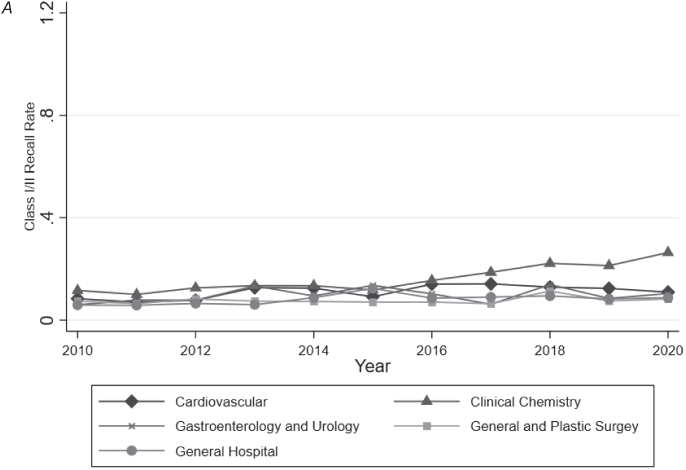
A: No software.

B: Software.
Note: Authors’ analysis of FDA approval and clearance data and the FDA’s MAUDE database from 2010 to 2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each year–specialty observation, total class I/II recalls were calculated and then divided by the number of approvals and clearances within that specialty in the preceding two years.
Figure 9.3 Two-year class I and class II recall rates by specialty over time.
Table 9.1 Two-year adverse event rates by specialty
| Specialty | Statistic | No software | Software | p |
|---|---|---|---|---|
| Cardiovascular | N | 3,055 | 1,341 | |
| Mean | 8.998 | 10.247 | 0.723 | |
| (SD) | (97.243) | (111.656) | ||
| Clinical chemistry | N | 1,067 | 332 | |
| Mean | 0.384 | 67.744 | 0.050 | |
| (SD) | (3.786) | (622.820) | ||
| Gastroenterology and urology | N | 1,530 | 329 | |
| Mean | 1.548 | 5.991 | 0.108 | |
| (SD) | (13.286) | (49.618) | ||
| General hospital | N | 2,214 | 263 | |
| Mean | 0.745 | 10.989 | 0.047 | |
| (SD) | (8.197) | (83.094) | ||
| General and plastic surgery | N | 2,424 | 631 | |
| Mean | 1.791 | 1.498 | 0.486 | |
| (SD) | (16.036) | (6.694) | ||
| Total | N | 10,290 | 2,896 | |
| Mean | 3.524 | 14.516 | 0.010 | |
| (SD) | (54.059) | (226.749) |
Note: Authors’ analysis of the FDA’s MAUDE and recall databases for devices approved/cleared from 2008 to 2018. Software identified based on keyword searches of FDA approval/clearance documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). Adverse events limited to mandatory reports. For each device, the total number of adverse events in two years following regulatory approval or clearance was calculated. Differences in means within specialties by software presence were assessed using two-sided t-tests under the assumption of unequal variance.
While devices with software generally experienced more adverse events and recalls, we observed significant heterogeneity in these differences by medical specialty area. When examining adverse events within individual medical specialties, only clinical chemistry and general hospital devices had statistically significant differences in adverse event rates in SdMDs vs. other devices. Among clinical chemistry devices, SdMDs had a mean 67.744 associated adverse events reported in the two years following regulatory approval or clearance, while non-SdMDs had a mean of just 0.384 adverse events reported in the two years following regulatory approval or clearance (p = 0.050) (Table 9.1). The difference between SdMDs and non-SdMDs, while statistically significant, was smaller among general hospital devices, where SdMDs had a mean of 10.989 associated adverse events in the two years following regulatory approval/clearance, while non-SdMDs had a mean of 0.745 adverse events reported over the same window of time (p = 0.047) (Table 9.1).
In contrast to adverse events, we observed significant differences in the number of recalls per approved device between SdMDs and non-SdMDs in each medical specialty studied. However, here too, the magnitude of the difference in recall rates varied meaningfully by specialty. General and plastic surgery devices had the smallest differences in recall rates (5.2 percent for SdMDs vs. 3.1 percent for non-SdMDs) (p = 0.025) (Table 9.2). General hospital devices had the largest difference in recall rates (11.8 percent of SdMDs vs. just 2.4 percent of non-SdMDs) (p < 0.001) (Table 9.2).
Table 9.2 Two-year class I and class II recall rates by specialty
| Specialty | Statistic | No software | Software | p |
|---|---|---|---|---|
| Cardiovascular | N | 3,055 | 1,341 | |
| Mean | 0.050 | 0.080 | <0.001 | |
| (SD) | (0.219) | (0.271) | ||
| Clinical chemistry | N | 1,067 | 332 | |
| Mean | 0.028 | 0.093 | <0.001 | |
| (SD) | (0.165) | (0.291) | ||
| Gastroenterology and urology | N | 1,530 | 329 | |
| Mean | 0.041 | 0.097 | 0.001 | |
| (SD) | (0.199) | (0.297) | ||
| General hospital | N | 2,214 | 263 | |
| Mean | 0.024 | 0.118 | <0.001 | |
| (SD) | (0.153) | (0.323) | ||
| General and plastic surgery | N | 2,424 | 631 | |
| Mean | 0.031 | 0.052 | 0.025 | |
| (SD) | (0.173) | (0.223) | ||
| Total | N | 10,290 | 2,896 | |
| Mean | 0.036 | 0.081 | <0.001 | |
| (SD) | (0.187) | (0.273) |
Note: Authors’ analysis of the FDA’s MAUDE and recall databases for devices approved/cleared from 2008 to 2018. Software identified based on keyword searches of FDA approval/clearance documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each device, a binary indicator for a class I or class II recall was calculated. Differences in means within specialties by software presence were assessed using two-sided t-tests under the assumption of unequal variance.
We also observed that the differences in outcomes between SdMDs and non-SdMDs were driven in part by large increases in recalls and adverse events for specific types of devices over relatively short periods of time. For example, a large increase in recalls of general hospital devices between 2011 and 2013 was primarily driven by recalls of infusion pumps and sterilizers. A large increase in recalls of clinical chemistry devices in 2018 through 2020 was primarily driven by recalls of blood glucose monitors. Table 9.3 presents illustrative examples of such recalls.Footnote 23
Table 9.3 Example recalls
| Infusion pump recall description: | Glucose monitor recall description: |
|---|---|
| “Moog Inc. … announced today that the [FDA] has classified the voluntary correction of the Curlin 6000 CMS, Curlin 6000 CMS IOD, PainSmart, and PainSmart IOD as a Class I recall… The decision to conduct the device recall is due to a software anomaly which leads to software Error Code 45 (EC45), resulting in a shutdown of the pump. This failure may result in a delay or interruption of therapy, which could result in serious injury and/or death.” | “… Dexcom… issued a voluntary recall on the G6 CGM App due to the alarm feature on the iOS application failing to properly alert users. In particular, alarms were not detecting severe hypoglycemic (low glucose) or hyperglycemic (high glucose) events and therefore consumers were not being notified of fluctuations to blood glucose levels.” |
V Discussion
Overall, we observed that SdMDs had higher adverse event and recall probabilities compared to devices without software components. Further, we documented heterogeneity in the difference between SdMDs and non-SdMDs, both over time and across medical specialties.
It should be noted that there are several limitations on the current post-market surveillance system in the United States that prevent us from concluding that SdMDs are less safe than non-SdMDs. For example, even if SdMDs experience more recalls and adverse events, software-based recalls may have a smaller impact on patient wellbeing vs. other types of recalls. For example, manufacturers may be able to address (some) software recalls more quickly by issuing software patches, rather than physically removing defective products from the market. However, in supplemental analyses (not reported here), we found no evidence that recalls of SdMDs were terminated more quickly (on average) than those of non-SdMDs.
In addition to limitations in our ability to extrapolate patient impact from adverse event and recall-based measures, there is almost certainly imprecision in how we estimated the rates of these outcomes. The FDA’s MAUDE database for reporting adverse events does not include the number of devices in use at any given time – that is to say, there is no “denominator” to calculate the frequency of adverse events and/or recalls per device in circulation. As such, it is impossible to calculate a true adverse event rate, defined as adverse events per medical device in use. Rather, we calculate the rates of adverse events and recalls per device approved, but this is an imperfect measure. Devices with more units in circulation may have had more adverse events simply because they were used in more patients, which in turn, could impact the interpretation of our findings. Specifically, if SdMDs were used more (or less) frequently than non-SdMDs, the true per device used probability of such events could be substantially lower (or higher, respectively).
Further, both adverse event reporting and recalls rely on users and manufacturers identifying product problems. The salience of product issues is therefore likely to influence the probability with which true product failures are reported as adverse events and result in product recalls. One could imagine that certain types of product issues may be more noticeable in SdMDs – for example, issues with a digital display or internet connectivity. To the extent that this is true, it could also influence the results reported here and would drive up the likelihood that adverse events associated with SdMDs are reported and, as a corollary, the likelihood that a manufacturer recall is issued.
Our findings, therefore, also speak to the limitations of the current post-market surveillance and adverse event reporting infrastructure in the United States. While we found that on a per-new-device basis, SdMDs were more likely to experience recalls compared to non-SdMDs, we did not always detect differences in adverse events between SdMDs and non-SdMDs. Adverse events are a noisy signal of post-market safety and are not necessarily a reliable predictor of subsequent medical device recalls. The user-reported nature of the information collected in MAUDE may limit its ability to detect unsafe products, as regulators have already acknowledged.
Precisely because of these limitations, we believe that a key policy recommendation from our findings is the need for the systematic collection of unbiased data describing the post-market performance of both medical devices and digital diagnostics specifically. The FDA, the Centers for Medicare and Medicaid Services, and other bodies should work to include standardized medical device identifiers in administrative claims data (i.e., records of provider services reimbursed by health insurers).Footnote 24 Doing so would allow researchers and regulators to reliably track the use of SdMDs and their subsequent outcomes, thus differentiating safety issues from data artifacts caused by differences in device circulation.
It may also be beneficial for the FDA to consider implementing a broader and more robust set of post-market surveillance activities as software becomes increasingly integrated into medical devices and diagnostic technologies. Such activities could involve more direct evaluations of safety. For example, the FDA could potentially initiate periodic audits of randomly selected SdMDs to ensure that devices are performing as intended.
However, future post-market surveillance initiatives need not necessarily involve data collection by the FDA. The digitization of medical devices may raise safety issues, but it also presents new opportunities to collect data on device use and safety. SdMDs intrinsically generate “digital exhaust,” or metadata through their use. Regulators should consider how they might encourage manufacturers to leverage such data (including data on frequency and duration of device use) as part of post-market surveillance strategies, potentially by tying pre-market approval to a clear post-market data monitoring plan when appropriate.
The FDA alone will not be able to execute some of these changes. As the FDA acknowledged in a recent report, the “faster cycles of innovation and the speed of change for medical device software would benefit from a new regulatory approach,”Footnote 25 but the FDA is constrained in the actions it can currently take. The scope of the FDA’s regulatory activities is largely determined by the original 1976 legislation that gave the agency the authority to regulate devices. New legislative authority is needed for the FDA to design regulatory approaches that best address the unique nature of medical device software.Footnote 26
As the FDA considers new regulatory approaches to SdMDs, patients and providers should be aware that the introduction of software into previously analog devices may present new safety concerns. These concerns will not always be readily identifiable through existing post-market surveillance mechanisms. Accordingly, health care providers should consider how they might “monitor the monitors” and ensure that newly adopted remote patient monitoring technologies work as intended.
VI Conclusion
In an analysis focusing on five key medical specialties and using over a decade of data, we found that medical devices with software components had more adverse events and recalls (per new device) as compared to devices without software. While these findings hint at potential safety challenges associated with SdMDs, the data available do not allow us to extrapolate further and calculate safety issues per device in circulation, a measure that would be more appropriate for informing individual patient/provider safety concerns. That said, the data analyzed here demonstrate that it is vital to continue to monitor the safety and effectiveness of SdMDs going forward. Further, patients and providers should not assume that existing post-market surveillance mechanisms are sufficient for detecting safety concerns in the early years following market entry for new products with software components.









 Open access
Open access