


1. Introduction
An ancestral polymorphism is defined as a polymorphism that originated as a result of mutation prior to the speciation event that generated the species in which it segregates. The presence of ancestral polymorphisms within a species, and their fixation subsequent to speciation, can contribute to divergence from a closely related species; this influences estimates of rates of sequence evolution, and may also lead to incorrect inferences concerning phylogenetic relationships (e.g. Gillespie & Langley, Reference Gillespie and Langley1979; Clark, Reference Clark1997; Maddison, Reference Maddison1997; Arbogast et al., Reference Arbogast, Edwards, Wakeley, Beerli and Slowinski2002; Hudson & Coyne, Reference Hudson and Coyne2002; McVicker et al., Reference McVicker, Gordon, Davis and Green2009; Cutter & Choi, Reference Cutter and Choi2010). In addition, estimates of the abundance of ancestral polymorphisms provide a test for balancing selection, since an excess frequency of ancestral polymorphisms within a gene or genetic region, relative to the level that would be expected under neutrality, is a signature of long-term balancing selection (Wiuf et al., Reference Wiuf, Zhao, Innan and Nordborg2004; Asthana et al., Reference Asthana, Schmidt and Sunyaev2005). For the purpose of interpreting the phylogenetic relationships of closely related species, and analysing the causes of variability within species, it is thus important to take into account the contribution of ancestral polymorphisms to variability within species and divergence between species.
Here, we extend a method for estimation of the proportion of ancestral polymorphisms among all polymorphisms within species, based on a comparison of three species, which was first introduced by Ramos-Onsins et al. (Reference Ramos-Onsins, Stranger, Mitchell-Olds and Aguadé2004) and subsequently elaborated by Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) . We apply it to a dataset of nearly 70 protein-coding genes for which DNA sequence polymorphism data are available in both Drosophila pseudoobscura and its close relative Drosophila miranda, using their relative Drosophila affinis as an outgroup (Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010), in an attempt to estimate the true level of ancestral polymorphism for these two species. We show that a substantial proportion of the synonymous variants in these two species are ancestral, and that a small number of genes with unusually high sequence diversity seem to show evidence of an excess of ancestral polymorphisms, suggestive of balancing selection. Our methods offer a substantial improvement on those reported in Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), by introducing novel procedures for the estimation of the parameters of interest. We also incorporate the estimation of confidence intervals on these parameters, in order to assess error in our estimates. In addition, we analyse a much larger dataset than Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) (67 genes compared with three genes), which enables us to compare levels of ancestral polymorphism at X-linked and autosomal loci.
2. Materials and methods
(i) Theoretical background
The method uses an outgroup species and parsimony to infer the ancestral state of a given polymorphic site in two species for which DNA sequence polymorphism data are available (Ramos-Onsins et al., Reference Ramos-Onsins, Stranger, Mitchell-Olds and Aguadé2004; Charlesworth et al., Reference Charlesworth, Bartolomé and Noël2005). The states of a given nucleotide site at the internal nodes of the phylogeny of that site are inferred from the observed state of the nucleotide in the outgroup species, from which a single DNA sequence is assumed to have been obtained. Thus, with three species denoted by X, Y and Z, where X and Y are close relatives for which polymorphism data are available and Z is the outgroup species, the state of a given nucleotide site in the outgroup is assumed to be the ancestral state for polymorphic sites in X and Y (see Fig. 1). In such a three-species comparison, the observed pattern of polymorphism at a nucleotide site across the three species can be assigned a ‘type’ that is consistent with the most parsimonious interpretation of the pattern (Charlesworth et al., Reference Charlesworth, Bartolomé and Noël2005).

Fig. 1. Interpretation of polymorphism patterns for a three-species model, using parsimony. Observed states are shown in blue, inferred states in green and inferred evolutionary events in red. Type 1 (a), type 2/3 (b) and type 4/5 (c) all represent ancestral polymorphisms, whereas type 6 (d) represents de novo polymorphisms that have arisen in one lineage of the tree. However, since type 4/5 is indistinguishable from type 6, they do not contribute to the observed fraction of ancestral polymorphisms. Adapted from Fig. 1 of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) .
Figure 1 displays an example of a C, T polymorphism observed at a given polymorphic site in a focal species (X) in a group of three species; the following arguments are equally true for polymorphisms observed in species Y, interchanging X and Y. Slightly modifying the terminology of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), we can define four distinct types of event that generate polymorphisms in species X: type 1, type 2/3, type 4/5 and type 6. Figure 1(a) shows a ‘type 1’ event: a CT polymorphism is observed in both species, while a T is present in the outgroup sequence. The most parsimonious interpretation is that the ancestral state for all three species was T, and that a T→C mutation occurred in the lineage leading to both species X and Y. In Fig. 1(b), a CT polymorphism is observed only in X, while Y is apparently fixed for C and the outgroup is T. Here, the most parsimonious explanation is that a T→C mutation that occurred in the common ancestor to X and Y gave rise to a CT polymorphism in both species, but in species Y either the C allele has gone to fixation (a ‘type 2’ event) or by chance the T allele was not found in the sample (a ‘type 3’ event). Although there is a clear distinction between a type 2 and a type 3 event, they are observationally identical, and both represent an ancestral polymorphism; they are thus pooled to constitute a ‘type 2/3’ event.
In Fig. 1(c), there is a CT polymorphism in species X, but only T is found in species Y and the outgroup. One possibility is that a T→C mutation occurred in the ancestral population prior to the speciation of X and Y, but the C variant has been lost from species Y (a ‘type 4’ event) or is not present in the sample taken from Y (a ‘type 5’ event). Alternatively, a de novo polymorphism that arose only in species X could have produced this pattern (a ‘type 6’ event: Fig. 1(d)). Type 4, 5 and 6 events are observationally indistinguishable, but have distinct evolutionary causes: type 4/5 events are ancestral polymorphisms, but cannot be distinguished from a ‘de novo’ polymorphism (type 6). Thus, it is this misclassification of type 4/5 polymorphisms as de novo (i.e. type 6) that constitutes the primary source of error in calculating the observed fraction of ancestral polymorphisms, which in its true sense is defined as the ratio of the sum of types 1 through to 5 to the total number of polymorphisms in a given species.
In order to estimate the fraction of ancestral polymorphisms among all polymorphisms in species X, we use the formulae of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) for calculating the expected frequencies of types 1, 2 and 3 events among the total, on the assumption of selective neutrality. Let Pd be the probability that a polymorphic site, which was present in the common ancestor of species X and Y, is classed as type 1 or type 2/3 (i.e. as an observed ancestral polymorphism) in species X. Let the probability of detecting a type i polymorphism in species X be denoted by Pi . For i=1–3, expressions for these probabilities are given by eqns (5), (6) and (9), respectively, of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), and can be summed to give Pd (eqn (11 a) of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) :

where n is the sample size for species Y, and t is the time since the split of the two species in question, measured in units of 2N e generations (here, N e is the effective population size for the lineage leading to the species designated as species Y, i.e. the non-focal species).
In order to use this result, an estimate of t is required. Using the expressions for the Pi in Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), we can equate the following functions of the observed and theoretical frequencies of types 1, 2 and 3 polymorphisms:

where f 1 is the observed fraction of type 1 polymorphisms and f [2+3] denotes the observed fraction of type 2/3 polymorphisms in species X. This provides a convenient exact formula for estimating t, which is more accurate than the approximate eqn (13) of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) .
Let the observed value of the expression on the left-hand side be denoted by r OBS; this can be equated to the relatively simple theoretical formula on the right-hand side, in order to obtain an estimate of t. A simple Java program (EstimateT; available on request) utilizes the Newton–Raphson method for solving a non-linear equation of the form f(x)=0, by iterating xi +1=xi −f(xi )/f′(xi ), where f(x) is a function of x and f′(x) is its derivative. In the present case, replacing x with t, the function that yields the desired estimate of t can be written as
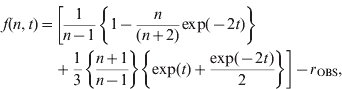
The partial derivative of f with respect to t is

Iterations using these expressions quickly yield a stable estimate of t for given values of r OBS and n. The method can be applied either to individual loci or a group of loci.
Following Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), the observed frequency of type 1/2/3 polymorphisms among all polymorphisms can then be divided by Pd , in order to correct for the misclassification of type 4/5 ancestral polymorphisms as type 6, yielding the estimated fraction of ancestral polymorphisms as r T. With independence among sites, this procedure is equivalent to a maximum likelihood estimate (see Supplementary material), assuming independence (linkage equilibrium) among nucleotide sites. While the assumption of linkage equilibrium is not completely accurate, polymorphism data show that linkage disequilibrium in these species falls off rapidly with distance between nucleotide sites (Schaeffer & Miller, Reference Schaeffer and Miller1993; Bachtrog & Andolfatto, Reference Bachtrog and Andolfatto2006), so that it is unlikely that it will pose a major problem in the case of these data. A possible effect of non-independence was tested for using the distribution across loci of the numbers of type 2/3 polymorphisms for the X chromosome and autosome of D. pseudoobscura, the only cases in which there is more than a handful of loci with more than one putatively ancestral polymorphism (see Supplementary Table S1). The mean numbers of type 2/3 polymorphisms per locus were 0·79 and 1·06 for the X chromosome and autosome, respectively; chi-squared tests for agreement with the Poisson distribution gave values of 6·29 and 3·27, respectively (3 df for each, P>0·05). Thus, there is no evidence for a non-random distribution across loci.
The variances and standard errors of t, Pd and r T can be calculated using the delta method (Bulmer, Reference Bulmer1980, p. 83), again assuming independence among sites so that the numbers of type 1, type 2/3 and type 4/5 and 6 polymorphisms are multinomially distributed (see Supplementary material). Alternatively, approximate confidence intervals for the estimates can be derived by bootstrapping. The dataset for the focal species is resampled (with replacement) k times, where k is equal to the number of polymorphic sites within the species, by randomly drawing a site from the array of sites from 1 to k, and storing it in a new array. This procedure is repeated 10 000 times; for each replicate, the f 1, f [2+3], fde novo (defined as the observed fraction of type 4/5 and 6 polymorphisms), t, Pd , r OBS and r T statistics are recalculated to create their sampling distributions; approximate 95% confidence intervals are then derived by extracting the 2·5 and 97·5 percentile values from these distributions.
(ii) Nature of the data
Our study species are D. pseudoobscura and D. miranda, with D. affinis as the outgroup species, as described by Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005) . D. pseudoobscura and D. miranda are very closely related, with a mean synonymous site divergence (K S) of about 4% (Bartolomé & Charlesworth, Reference Bartolomé and Charlesworth2006; Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010). Introgression between the two species is thought to be absent in the wild, and laboratory hybrids are completely infertile (Dobzhansky & Tan, Reference Dobzhansky and Tan1936), so that we should be safe to assume that the pattern of polymorphism observed here is not due to ongoing introgression between these species. This is important as ongoing hybridization would leave a pattern similar to that of ancestral polymorphism. D. affinis is another North American species that is relatively distantly related to our ingroup species, with a mean K S of about 25% for the X chromosome and 28% for the autosome (A) (Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010). The relatively large distance to the outgroup species poses some problems for the parsimony models used here, which are considered in section 2 (iii) below.
To estimate the incidence of ancestral polymorphism for D. pseudoobscura and D. miranda, the 67 loci that did not depart significantly from neutrality on the basis of a multilocus Hudson–Kreitman–Aguadé (HKA) test (Hudson et al., Reference Hudson, Kreitman and Aguadé1987; Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010) were screened for the presence of type 1, type 2/3 and type 4/5 and 6 synonymous polymorphisms in each species, using D. affinis as an outgroup. Gene sequence alignments for 34 autosomal (Muller element B or chromosome 4 in D. pseudoobscura) and 33 X-linked (Muller element A) loci that are orthologous in all three species were obtained (for details concerning Muller's elements, see Ashburner et al., Reference Ashburner, Golic and Hawley2005). Each alignment consisted of 12–16 sequences from both D. pseudoobscura and D. miranda, and one sequence from the outgroup species D. affinis. A polymorphism dataset was constructed for each alignment using the relevant functions in the software package DnaSPv5 (Librado & Rozas, Reference Librado and Rozas2009).
For all analyses, only polymorphisms at synonymous sites were used, as these are likely to be closer to neutrality than non-synonymous changes. Any alignment gaps were also excluded from the analysis. Some additional autosomal and X-linked loci that were previously identified as potentially being under selection in either D. pseudoobscura or D. miranda on the basis of the HKA test (Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010), and that were excluded from the main results presented here, are considered in the Discussion section.
A second Java program (PolyFinder; available on request) was written, which detects and classifies each type of polymorphism as a type 1, type 2/3 or type 4/5 and 6 for each species. When applied to the several hundred polymorphic sites in the samples from the two species, this program provides a simple and effective way of classifying polymorphisms under the parsimony assumption.
(iii) Corrections for errors in the parsimony inferences
A method of correcting errors in inferences from parsimony was described in the Appendix of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), and was applied to the present dataset. This method requires estimates of the numbers of polymorphisms involving transitions and transversions, respectively; the relevant data are provided in Table S2 of the Supplementary material. We also require an estimate of the time since the divergence of D. pseudoobscura and D. miranda to initiate the parsimony-correction procedure, because it requires use of the estimates of P 2, P 3 and the a priori probability of an ancestral polymorphism, but we also need accurate values for the proportions of type 1 and type 2/3 polymorphisms to estimate t from eqns (3) and (4). The correction procedure was performed on a locus-by-locus basis, thereby taking into account the slight variation in sample size between genes.
For each of the branches leading to D. pseudoobscura and D. miranda from their common ancestor, we therefore calculated a parsimony-free initial estimate of the time since divergence, t 0, using the ratio of the synonymous divergence between the species in question (K S) to the mean synonymous diversity (πS) of the non-focal species; this is expressed on a time-scale of units of 2N e generations, where N e is the effective population size along the lineage in question, which we equated to the estimate of current effective size for the species (Hudson et al., Reference Hudson, Kreitman and Aguadé1987). These values were 2·72 and 2·24 for the X and A of D. pseudoobscura, and 10·3 and 9·43 for the X and A of D. miranda, respectively. The initial estimate for a given chromosome and focal species was then used to calculate the proportion of incorrect assignments, as described by Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), and the corrected values were then used to recalculate t via the Newton–Raphson method outlined in section 2(i). No further use was made of the divergence/diversity ratios in subsequent iterations. Iterations were carried out until estimates of both t and the corrected values of f 1 and f [2+3] converged to three decimal places.
3. Results
(i) Frequencies of the different types of polymorphisms
The observed counts of ancestral polymorphisms for the autosomal and X-linked genes in D. pseudoobscura and D. miranda are shown in Table 1 (for a locus-by-locus breakdown of all polymorphisms, see Table S1 of the Supplementary material). These counts represent the observed values prior to correction of errors in the parsimony assumptions used in their detection. The observed fraction of apparent ancestral polymorphisms is the sum of the type 1 and type 2/3 polymorphisms, divided by the total number of polymorphisms. These account for (10+62)/528=0·136 and (10+15)/128=0·195 of the total polymorphisms seen within D. pseudoobscura and D. miranda, respectively. As discussed previously, this observed fraction is likely to be biased in two ways, firstly, by the misclassification of 4/5 polymorphisms as de novo polymorphisms and secondly by errors in the parsimony methodology used to classify polymorphisms within this dataset. To deal with these problems, an estimate of divergence time between the two species is required, as described in section 2.
Table 1. Numbers of different types of polymorphisms found in D. pseudoobscura and D. miranda, before correcting for parsimony errors

A and X refer to autosomal and X-linked loci, respectively.
k is the number of loci in each category.
(ii) Divergence times and corrections for parsimony error
The estimates of the number of different types of polymorphisms after correction for parsimony errors, using the method outlined in section 2(iii), are shown in Table 2. The corrections reduce the number of observed ancestral polymorphisms (i.e. type 1/2/3) from 72 to ~33 (for D. pseudoobscura) and from 25 to ~15 (D. miranda). Thus, correcting for parsimony errors reduces the estimate of the proportion of type 1/2/3 polymorphisms by approximately half.
Table 2. Number of different types of polymorphisms found in D. pseudoobscura and D. miranda, after correcting for parsimony errors

A and X refer to autosomal and X-linked loci, respectively.
Estimates of the time since the divergence of D. pseudoobscura and D. miranda, after corrections for parsimony errors, were obtained as described in sections 2(i) and 2(iii), and are shown in Table 3. These divergence time estimates are in units of 2N e generations, where N e is the long-term effective population size of the lineage leading to the species chosen as Y in the comparisons, i.e. the partner to the focal species whose polymorphism data are being used (see section 2(i)). The estimated times for both the X chromosome and autosome are greater than 1, so that the use of the equations in section 2(i) is justified, since these require t>0·5 (Charlesworth et al., Reference Charlesworth, Bartolomé and Noël2005).
Table 3. Estimates of t, Pd and r T after correcting for parsimony errors

Estimates are presented with standard errors in parentheses and 95% confidence intervals below (based on 10 000 replicate populations of the corrected data; see section 2(ii)). Time t is in units of 2N e generations. Pd is the probability of classifying an ancestral polymorphism as such, r T is the true fraction of ancestral polymorphisms; both are expressed as percentages.
A and X refer to autosomal and X-linked loci, respectively.
The estimates of Pd , defined by eqn (1) of section 2(i), are all around 40%, indicating that there is a high probability of misclassification of an ancestral polymorphism. As described in section 2(i), this allows estimation of the fraction of ancestral polymorphisms by dividing the frequency of type 1/2/3 polymorphisms by the estimate of Pd . The results show that there is a significant frequency of ancestral polymorphisms on both the X chromosome and autosome in both species, which is higher for the X (especially in D. miranda), and higher in D. miranda than in D. pseudoobscura.
This method also offers a way of calculating the relative proportions of the indistinguishable type 4/5 (ancestral) and type 6 (true de novo) polymorphisms; the difference between the unadjusted and adjusted fraction of ancestral polymorphisms is attributable to the misclassified polymorphisms that were erroneously categorized as de novo. Since these are type 4/5 polymorphisms, this difference must be equal to the total proportion of type 4/5 polymorphisms in a given dataset. Thus, for the X chromosome in D. pseudoobscura, the (corrected) proportion of type 1/2/3 polymorphisms is 7·7% and the estimated proportion of ancestral polymorphisms is 19·5%, so that the estimate of the proportion of type 4/5 polymorphisms is 11·8%. It is interesting to note, therefore, that type 4/5 polymorphisms appear to contribute more to the overall fraction of ancestral polymorphisms than type 1 and type 2/3 combined. This reflects the fact that, as may be seen from Fig. 1, type 4/5 events involve the loss of the derived rather than ancestral variant from either the population or the sample; since derived variants are mostly present at low frequencies, their loss is more probable than the loss of the ancestral variant.
4. Discussion
The results presented here suggest that a significant fraction of polymorphisms in D. pseudoobscura and D. miranda arose in the ancestral population common to these species, prior to their complete separation by speciation. We estimate that the overall proportions of ancestral polymorphisms are 19·5±4·9% (standard error) and 14·9±3·8% for the D. pseudoobscura X chromosome and autosome, respectively; the corresponding values for D. miranda are 47·6±14·1% and 10·0±6·4%. Our result for the D. pseudoobscura autosome agrees well with a previous estimate of 18·6% (Charlesworth et al., Reference Charlesworth, Bartolomé and Noël2005), but is likely to have a substantially lower standard error due to the increased size of the present dataset.
The main novelty of the approach used here is that it enables an adjustment to be made for the ancestral polymorphisms that are undetected in the sample (the type 4/5 polymorphisms; see section 2(i)), as well as using parsimony to detect polymorphisms that are ancestral but not shared between the two species in question (type 2/3 polymorphisms, shared polymorphisms being classed as type 1). Before this adjustment, but after correcting for parsimony error, the estimated fractions of ancestral polymorphisms were approximately 6% and 12% for D. pseudoobscura and D. miranda, respectively (Table 2). The adjustment therefore increases the estimated fraction of ancestral polymorphisms by a factor of more than two. The parsimony correction used here is, however, somewhat crude (for details, see Charlesworth et al., Reference Charlesworth, Bartolomé and Noël2005), and so the numerical results should be treated with caution. Our method of adjusting for misclassified ancestral polymorphisms would be more trustworthy when used with an outgroup species with a much lower divergence than that between D. affinis and the two focal species, so that no parsimony correction is required, but no such species is currently available.
It is clear, however, that it is not sufficient to infer the level of ancestral polymorphisms on the basis of the number of shared polymorphisms alone, as has been done in most previous studies. This underestimates the total number of ancestral polymorphisms by a factor of about two, which may bias estimates of sequence divergence (Gillespie & Langley, Reference Gillespie and Langley1979; Patterson et al., Reference Patterson, Richter, Gnerre, Lander and Reich2006; McVicker et al., Reference McVicker, Gordon, Davis and Green2009; Cutter & Choi, Reference Cutter and Choi2010). In addition, use of the fraction of unequivocally identified shared polymorphisms alone may underestimate the role of balancing selection, which has previously been reported to be limited in a human–chimpanzee study of shared polymorphisms (Asthana et al., Reference Asthana, Schmidt and Sunyaev2005).
The estimated divergence time for the autosome is greater than that in the case of the X chromosome in D. pseudoobscura, and the values with regard to D. miranda are both smaller than for D. pseudoobscura. These differences are in the opposite direction from what is expected based on the estimates of N e for these chromosomes and species, given current levels of synonymous nucleotide site diversities (Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010, Reference Haddrill, Zeng and Charlesworth2011). These indicate that both species have N e values for the X chromosome that do not differ significantly from three-quarters of that for the autosomes, as expected for species with little non-random variance in male mating success due to sexual selection (Charlesworth, Reference Charlesworth2009). In addition, D. miranda has about one-quarter the silent site diversity of D. pseudoobscura, so that its scaled divergence time should be four times that of D. pseudoobscura; the X chromosome in both species has about three-quarters of the diversity of the autosomes, so that its scaled divergence time should be four-thirds of that of the autosomes. While the X–A difference in D. pseudoobscura is not significant, the t values for D. miranda are highly significantly different from four times the D. pseudoobscura values, and are very different from the estimates based on the ratio of divergence to diversity given in section 2(iii). These discrepancies almost certainly reflect the fact that D. pseudoobscura has undergone a recent population expansion, whereas the unusually low diversity of D. miranda suggests that it may have experienced a past contraction of population size (Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010, Reference Haddrill, Zeng and Charlesworth2011). Thus, the long-term N e along the D. pseudoobscura lineage is probably much smaller, and the value for D. miranda much larger, than indicated by contemporary diversity estimates. While the t estimates in Table 3 are fairly noisy, it seems likely that they are less biased than the estimates from divergence/diversity ratios. Our estimates of the fraction of ancestral polymorphisms should not, however, be affected by shifts in population size along the D. pseudoobscura and D. miranda lineages, if we regard the effective sizes in the relevant equations as representing the harmonic mean effective sizes along the relevant lineage (Charlesworth & Charlesworth, Reference Charlesworth and Charlesworth2010, pp. 225–226).
In D. miranda, there is a large difference between the estimated proportion of ancestral polymorphisms on the X chromosome and the autosome, in the direction opposite to that expected from the difference in effective population sizes, and hence t, for these two chromosomes. From the expression given on p. 154 of Charlesworth et al. (Reference Charlesworth, Bartolomé and Noël2005), the expected ratio of X to A ancestral polymorphism levels is approximately equal to exp (t A−t X), where the subscripts denote the divergence time for the chromosome in question. If the estimated t values for D. miranda from Table 3 are substituted into this expression, the ratio of expected ancestral polymorphism levels is thus exp (−0·31)=0·73. More conservatively, if we use the lower confidence limit for t X (equal to 0·84) and estimate t A as three-quarters of this, we obtain a ratio of 0·81. By multiplying the observed proportion of autosomal ancestral polymorphisms by this ratio, we can adjust this proportion to a scale on which it can be compared with the fraction for the X chromosome.
Even if the more conservative estimate is used for this purpose, the confidence interval for the adjusted A value does not overlap with that for X (the upper limit for A is 21·6 compared with a lower limit for X of 23·2). This suggests that there may be a real difference between X and A, compared to what is expected on the model used to generate the predictions. A possible explanation for this is that a relatively recent reduction in effective population in the D. miranda lineage has had a greater effect on the X (with its lower effective population size) in causing loss of the most recent derived polymorphisms, leading to a deficiency of type 6 events (see Fig. 1).
Finally, we examine how ancestral polymorphism levels may relate to the expectation of an elevated synonymous site diversity associated with long-term balancing selection (for a review of the theory of this effect, see Charlesworth & Charlesworth, Reference Charlesworth and Charlesworth2010, pp. 393–398). Haddrill et al. (Reference Haddrill, Loewe and Charlesworth2010) found seven additional loci that appeared to show significantly elevated synonymous diversity in one or other of the two species, on the basis of a multilocus HKA test (Hudson et al., Reference Hudson, Kreitman and Aguadé1987); this implies that their elevated diversity cannot simply be a consequence of a randomly generated, long coalescence time and that a non-neutral process is likely to be needed to explain the properties of these genes. Table 4 shows the uncorrected estimates of the numbers of ancestral synonymous polymorphisms for these loci. Comparison with Table 1 suggests that there may indeed be an excess of type 1/2/3 polymorphisms when compared with loci that show no deviations from neutral expectations by the HKA test. This possibility can be tested using a 2×2 contingency table for ‘ancestral’ vs. ‘de novo’ polymorphisms for the two categories of loci. For the X chromosome, the 2×2 χ2 is 8·15 (P<0·01), and for the autosome (with much less data) it is 1·85 (non-significant). While this test is somewhat crude, it suggests that the possibility of balancing selection acting on at least some of these genes is worth further investigation.
Table 4. Uncorrected numbers of different types of polymorphisms found in D. pseudoobscura and D. miranda for three autosomal and five X-linked genes with high nucleotide diversity in at least one of the species

πs (%) pse/mir is the mean pairwise nucleotide diversity for D. pseudoobscura and D. miranda, respectively (from Table 2 of Haddrill et al., Reference Haddrill, Loewe and Charlesworth2010).
A and X refer to autosomal and X-linked loci, respectively.
% of type is the percentage of all polymorphisms of that type contributed by the specific genes listed.
In summary, we have extended and substantially improved a method for estimation of the proportion of ancestral polymorphisms within species by introducing novel procedures for estimation of the scaled divergence time between species, correcting for parsimony and generating confidence intervals on the parameters of interest. We have applied this improved method to a dataset of polymorphisms in almost 70 protein-coding genes, distributed across the X chromosome and an autosome in D. pseudoobscura and D. miranda, and find that a substantial proportion of synonymous variants in these two species are ancestral.
We thank Kai Zeng for assistance with the use of Java. R. N. was funded by a Masters Studentship from the Natural Environment Research Council, and P. H. is funded by a Natural Environment Research Council Fellowship. The data were generated with funding from a research grant to B. C. from the Natural Environment Research Council. We thank two reviewers for comments that helped improve this paper.
Supplementary material
The online data are available at http://journals.cambridge.org/GRH






