


Obesity is now one of the largest public health challenges( Reference Masters, Reither and Powers 1 ). Worldwide, the prevalence of obesity has nearly doubled since 1980( 2 ). In 2014, 11 % of men and 15 % of women were obese. In the USA, 67·3 % and 33·7 % of the adult population are overweight (BMI≥25 kg/m2) and obese (BMI≥30 kg/m2), respectively( 2 ). The secular trend in obesity has multiple causes, including genetics, exposure to environmental toxins, increased food availability, altered frequency of eating, increased energy density in foods, physical inactivity and socio-economic status( Reference Holtcamp 3 – Reference van Vliet-Ostaptchouk, Snieder and Lagou 5 ).
The volume of adipose tissue (AT) in obese individuals correlates with the increased levels of cytokines and chemokines such as TNF-α, IL-1 and IL-6, and acute-phase proteins such as C-reactive protein. The increased levels of pro-inflammatory cytokines in blood may originate from the AT( Reference Weisberg, McCann and Desai 6 , Reference You, Yang and Lyles 7 ). In obesity, the expansion of the AT is associated with a local infiltration of inflammatory cells, mainly macrophages that gives rise to low-level chronic inflammation( Reference Weisberg, McCann and Desai 6 , Reference Xu, Barnes and Yang 8 ). It is believed that a chronic low-grade inflammation in the AT in obese individuals contributes to the increased mortality and morbidity by increasing the risks from complications such as insulin resistance, type 2 diabetes, hypertension, the metabolic syndrome, coronary artery diseases, non-alcohol fatty liver disease and cancer( Reference Gualillo 9 – Reference Rask-Madsen and Kahn 13 ). Despite an intense research in AT inflammation, it has not yet been possible to find the factors that trigger and sustain the inflammation( Reference Giordano, Murano and Mondini 14 ). Nevertheless, certain AT are localised in close proximity to barriers to the external environment. The subcutaneous AT (SAT) and visceral AT (VAT) are exposed to microbial products( Reference Peyrin-Biroulet, Chamaillard and Gonzalez 15 , Reference Zhang, Guerrero-Juarez and Hata 16 ). Recently, we have described how lipopolysaccharide (LPS) from the gut may enhance the transport of lipids to the AT by facilitating transcytosis through the capillary endothelium( Reference Hersoug, Moller and Loft 17 ). The exposure of AT to LPS depends on the lipid content, the type of lipids in the diet, and the gut permeability to LPS( Reference Caesar, Tremaroli and Kovatcheva-Datchary 18 ). The latter is influenced by the microbiome and the composition of the ingested food( Reference Chakraborti 19 ). In the present review, we focus on associations between AT inflammation and exposure to LPS from the gut microbiota. We hypothesise that ingestion of lipids facilitates a translocation of LPS from the gut microbiome to the circulation. Furthermore, we hypothesise that LPS is involved in defining the adipocyte death size, formation of crown-like structures, activation of caspase-4/5/11, and induction of pyroptosis in adipocytes.
The present review is based on literature searches conducted using PubMed and Web of Science for full-text English language original research and review articles published from 1996 to June 2016 with the following key words and their combinations: obesity, adipose tissue, adipocytes, inflammation, gut microbiota, LPS, endotoxemia and inflammasome. Articles were included if they described causes of inflammation in AT, the role of the gut microbiota in AT inflammation, cellular reactions to LPS exposure and pyroptosis. Reference lists from the articles in the initial search were also assessed for relevant articles to include in the review.
Adipose tissue in obesity
AT plays several functions depending on its specialisation (brown or white AT) and location (SAT or VAT). It secretes a number of signalling molecules (for example, chemokines, interleukins and adipokines) that regulate energy homeostasis, innate immunity and inflammation( Reference Galic, Oakhill and Steinberg 20 ). AT contains pre-adipocytes, fibroblasts, endothelial cells, immune cells and multi-potent stem cells. The cells are embedded in the extracellular matrix, which is composed of collagen 1, 3, 4 and 6, different proteoglycans, and matrix metalloproteinases( Reference Divoux and Clement 21 , Reference Khan, Muise and Iyengar 22 ). The volume of AT can increase by increasing the volume of pre-existing adipocytes (hypertrophy) or by generating new small adipocytes (hyperplasia). The expansion is caused by an increased rate of TAG storage (lipogenesis) and a decreased TAG removal rate (lipolysis)( Reference Arner 23 ). Increased size of adipocytes within the AT correlates with insulin concentration, insulin resistance and increased risk of developing type 2 diabetes( Reference Weyer, Foley and Bogardus 24 – Reference Krotkiewski, Bjorntorp and Sjostrom 26 ). Adipocyte hypertrophy may impair AT function( Reference Arner, Westermark and Spalding 27 ). AT with low recruitment rates of small pre-adipocytes displays hypertrophy, whereas a high rate of recruiting pre-adipocytes is associated with hyperplasia( Reference Arner, Westermark and Spalding 27 ). Subjects with AT hypertrophy have increased secretion of inflammatory proteins, enhanced lipolytic activity and decreased insulin-induced glucose metabolism in adipocytes( Reference Gao, Mejhert and Fretz 28 ). Therefore, hypertrophic AT is termed ‘unhealthy’, whereas hyperplasic AT is termed ‘healthy’( Reference Lessard, Laforest and Pelletier 29 ). Furthermore, it has been suggested that the unhealthy properties of hypertrophic AT is caused by a lower capacity to store lipids compared with hyperplasic AT( Reference Virtue and Vidal-Puig 30 ). This is associated with metabolic complications and insulin resistance due to ectopic deposition of lipids in non-AT( Reference Virtue and Vidal-Puig 30 ). Animal studies have shown that LPS exposure increases the proliferation of pre-adipocytes through a cluster of differentiation (CD) CD14-dependent mechanism, whereas CD14-positive cells are not recruited from non-adipose depot origin( Reference Luche, Cousin and Garidou 31 ). On the other hand, another study has shown that LPS exposure reduces adipogenesis in AT by disrupting the differentiation and inducing premature senescence of pre-adipocytes( Reference Zhao and Chen 32 ). Therefore, LPS can influence the balance between hypertrophia and hyperplasia in AT, but the overall effect may depend on concentration, genetic background and the local chemical environment. Time-course studies of the size and number of adipocytes in rats suggest that a ‘critical’ adipocyte size threshold triggers the recruitment of pre-adipocytes, which eventually differentiate into mature adipocytes( Reference Faust, Johnson and Stern 33 ). This may explain the interindividual differences in the adipocyte number associated with fat mass in adults(34). The numbers of adipocytes are remarkably constant in adulthood in both obese and lean individuals even after weight loss( Reference Spalding, Arner and Westermark 34 ). Adipocytes and macrophages share several characteristics related to the storing of lipids and regulation of metabolic homeostasis. In conditions such as overnutrition and obesity, pre-adipocytes act as immune cells, develop phagocytic and antimicrobial properties, and differentiate into macrophages( Reference Charriere, Cousin and Arnaud 35 , Reference Cousin, Munoz and Andre 36 ).
It has been shown that the adipocytes from VAT are smaller than those from SAT in the same subject( Reference Goldrick 37 , Reference Meyer, Ciaraldi and Henry 38 ). The adipocytes from VAT are also more metabolically active than their counterparts in SAT, whereas large adipocytes are more metabolically active and have a higher adipokine production( Reference Skurk, Alberti-Huber and Herder 39 ). In obesity VAT contains more macrophages as compared with SAT( Reference Weisberg, McCann and Desai 6 ). However, it is unresolved whether or not adipocytes in VAT and SAT have different responsiveness to LPS stimulation.
Cellular changes and formation of crown-like cells in adipose tissue during obesity
Macrophages were the first leucocytes to be identified in hypertrophic AT. Subsequently, natural killer cells, B lymphocytes, and CD4+ Th1 and CD8+ T lymphocytes have been found in AT and the number of these cells increase in obesity( Reference O’Rourke and Lumeng 40 ). Recent studies have demonstrated novel effector and regulatory roles of other cells in the innate and adaptive immune system, such as T regulatory (Treg) cells, neutrophils, eosinophils, mast cells and innate lymphoid type 2 cells( Reference Lee and Lee 41 ). In AT, macrophages can be activated by classic or alternative pathways (i.e. macrophage phenotype 1 (M1) or macrophage phenotype 2 (M2), respectively), depending on signals from the extracellular milieu( Reference Martinez, Sica and Mantovani 42 ). Macrophages can develop the M1 phenotype when exposed to microbial products or cytokines such as IL-4, IL-10, IL-13 and IL-33( Reference Locati, Mantovani and Sica 43 ). The M1 macrophages are characterised by an IL-12high, IL-23high and IL-10low phenotype( Reference Locati, Mantovani and Sica 43 ). Macrophages with the M1 phenotype participate as inducers in polarised Th1 responses, which are primarily directed against intracellular infections (cytosolic LPS), parasites and tumours. M2 cells are involved in tissue remodelling and express high levels of scavenger-, galactose- and mannose-type receptors( Reference Mantovani, Biswas and Galdiero 44 ). Interestingly, resident macrophages have a predominantly M2 phenotype in AT from lean subjects, whereas they display predominantly the M1 phenotype in obese subjects( Reference Harford, Reynolds and McGillicuddy 45 ). It has been shown that the number of M1 macrophages increase dramatically in obese mice on a high-fat diet (HFD), while the increase in the number of M2 macrophages is modest( Reference Fujisaka, Usui and Bukhari 46 ). This might be due to increased exposure to intracellular microbial cues, rather than by extracellular microbial cues, because the Th1 response primarily is directed against intracellular infections.
It has been shown that the rate of adipocyte death is increased by 30-fold with obesity in both mice and human subjects( Reference Cinti, Mitchell and Barbatelli 47 ). The presence of dead adipocyte remnants and macrophages gives rise to so-called crown-like structures( Reference Cinti, Mitchell and Barbatelli 47 ). The involvement of macrophages indicates that adipocytes die by pyroptosis, which is morphologically distinct from apoptosis because there is little formation of nuclear and mitochondrial degeneration( Reference Aki, Funakoshi and Uemura 48 ). Infiltration of macrophages into AT has clinical importance due to the association with the onset of insulin resistance( Reference Weisberg, McCann and Desai 6 , Reference Xu, Barnes and Yang 8 , Reference Schenk, Saberi and Olefsky 49 , Reference Zhang, Wright and Bernlohr 50 ). It is well known that dying cells have to be removed by macrophages, whereas the mechanism behind adipocyte cell death remains to be unravelled. In addition, it seems that the largest proportion of dying cells consist of large adipocytes( Reference Cinti, Mitchell and Barbatelli 47 ). We hypothesise that macrophages are recruited to the AT by adipocytes and they are subsequently transformed from M2 to M1 cells during obesity by microbial substances( Reference Hersoug, Moller and Loft 17 )(Fig. 1).
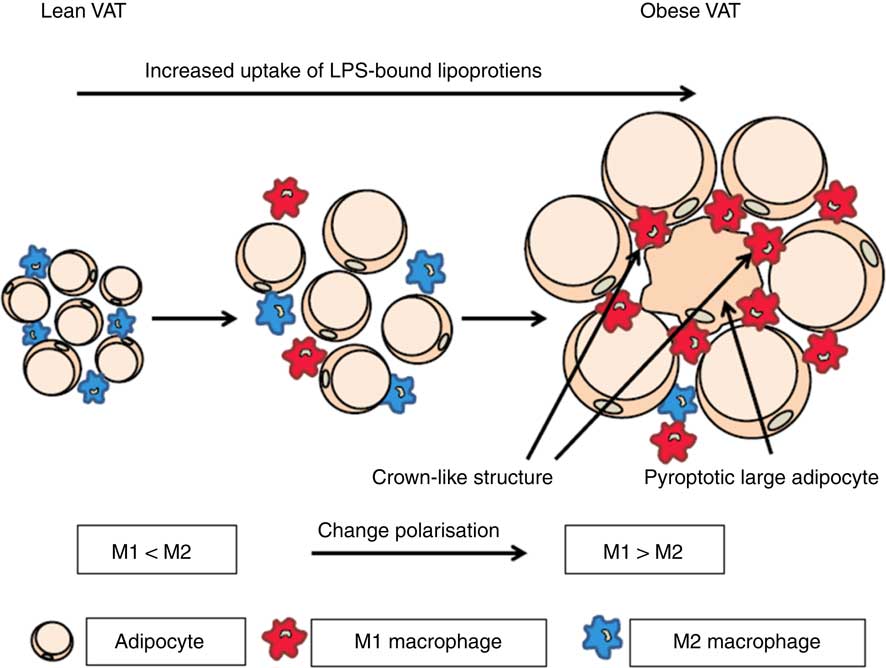
Fig. 1 Activation of macrophages in adipose tissue by microbial substances such as lipopolysaccharide (LPS) in lipoproteins. In lean visceral adipose tissue (VAT) the macrophages are predominately of the M2 phenotype. During transition towards obesity, adipocytes increase in size and the phenotype of macrophages changes to the inflammatory M1 phenotype and an increasing number of large adipocytes die of pyroptosis. When adipocytes enlarge they become increasingly metabolic active and internalise increasing quantities of lipoproteins with associated LPS. This increases the possibility of a non-canonical activation of caspase-4/5/11 leading to pyroptosis. For a colour figure, see the online version of the paper.
This hypothesis is supported by observations that obese subjects have higher plasma levels of LPS than do lean subjects( Reference Troseid, Nestvold and Rudi 51 ). In addition, it has been shown that the plasma levels of LPS have a stronger correlation with intra-abdominal fat volume than with subcutaneous fat volume. This was hypothesised to be related to a closer proximity of the VAT to the gut and a relatively higher exposure to bacterial components. The same study also showed that the microbial DNA content was successively higher in AT from subcutaneous, omental and mesenteric tissue samples( Reference Milinovich, Burrell and Pollitt 52 ).
Translocation of lipopolysaccharide from the gut and metabolic endotoxaemia
The diversity of the gut microbiota is huge as exemplified by observations of 100 times more bacterial genes than the entire mammalian genome( Reference Qin, Li and Raes 53 ). These bacteria usually colonise the gastrointestinal tract of the host without causing symptoms and they typically extend the capacity to degrade ingested foods, but they may also cause diseases( Reference Xu and Gordon 54 ). The extended metabolic capacity of the microbiota provides a supply of vitamins, antioxidants and SCFA( Reference Devkota, Wang and Musch 55 , Reference Lam, Su and Koprowski 56 ). The microbiota also releases LPS, which is composed of an O-antigen, a core region, and a lipid A tail (lipophilic lipid). LPS is an amphipathic molecule (a molecule having hydrophobic and hydrophilic regions). It is primarily the lipid A part that is responsible for the high toxicity and immunomodulatory property of LPS( Reference Santos, Silva and Castanho 57 ).
Ingestion of a HFD has been associated with elevated levels of Gram-positive (Firmicutes) and decreased levels of Gram-negative (with LPS in the cell wall) (Bacterioidetes). The Firmicutes:Bacterioidetes ratio is higher in heavily obese individuals compared with ‘healthy obese’ and lean individuals( Reference Furet, Kong and Tap 58 , Reference Louis, Tappu and Damms-Machado 59 ). Reduction in body weight as a consequence of an energy-restricted diet has been shown to reduce the Firmicutes:Bacterioidetes ratio to values that are similar to that in lean individuals( Reference Ley, Turnbaugh and Klein 60 ). Studies in both human subjects and mice have shown that ingestion of a HFD increases the proportion of Firmicutes. However, some human studies did not show an increased proportion of Firmicutes in obese individuals( Reference Duncan, Lobley and Holtrop 61 ) and some studies even showed a decreased proportion of Firmicutes( Reference Schwiertz, Taras and Schafer 62 ). An explanation of this discrepancy was recently suggested in a study using meta-genomics and microarray-based meta-transcriptomics analysis of stool samples. It showed multiple microbial community-wide transcriptional interactions and that gene regulation played an important role in bacterial species adaption and niche segregation in the gut( Reference Plichta, Juncker and Bertalan 63 ). Classic population ecology theory predicts that competition between two undifferentiated species leads to extinction of one species(64). This is not the case in the human gastrointestinal tract where many microbial species co-exist. One way to develop co-existence with other species could be through differentiation by transcriptional adaption( Reference Plichta, Juncker and Bertalan 63 ). This implies that the microbiome has this ability both quantitatively and qualitatively. It could be an explanation for the different observations of the abundance of Firmicutes in different studies. In addition, HFD have been associated with decreased levels of Gram-positive Bifidobacterium in the gut and elevated plasma LPS levels in mice( Reference Medzhitov and Horng 65 , Reference Taira, Yamaguchi and Shimizu 66 ). It has been shown that a HFD increases the plasma concentration of LPS by 2- to 3-fold as compared with a low-fat low-carbohydrate control diet and a high-carbohydrate diet( Reference Cani, Amar and Iglesias 67 ). Intestinotropic proglucagon-derived peptide (glucagon-like peptide-2; GLP-2) is produced by L cells in the intestinal mucosa and it promotes intestinal growth and barrier function via insulin-like growth factor-1 and β-catenin pathways. The decrease in Bifidobacterium results in decreased production of GLP-2, decreased gene expression levels of tight junction proteins zona occludens 1 and occludin, and increased gut permeability( Reference Cani, Possemiers and Van de Wiele 68 ). The species Akkermansia muciniphila, which colonises the mucus layer in the intestine, has been shown to induce the gene expression of zonula occludens 1 and occludin in ApoE knockout mice( Reference Li, Lin and Vanhoutte 69 ). A deficiency of A. muciniphila might be associated with reduced tightness of the gut mucosa and increased translocation of LPS to the circulation( Reference Hersoug, Moller and Loft 17 ). Moreover, the postprandial period may be protracted after intake of a HFD, leading to a higher rate of translocation of LPS( Reference Hersoug, Moller and Loft 17 ).
Dietary fat and its influence on the microbiota
Ingestion of lipids promotes the excretion of bile from the liver to the small intestine, which solubilises dietary lipids through the formation of micelles. SFA promote the hepatic production of taurine-conjugated bile acids, whereas an isoenergetic diet rich in PUFA (safflower-seed oil) and glycine-conjugated bile acid increases the production of glycine-conjugated bile acids( Reference Devkota, Wang and Musch 55 ). The primary bile acids in humans are cholic acid and chenodeoxycholic acid and these acids are transformed by the microbiota to deoxycholic acid and lithocholic acid, respectively( Reference Lefebvre, Cariou and Lien 70 ). Deconjugation of the taurine-conjugated bile acids stimulates the growth of Bilophilia wadsdorpia. Dietary cholic acid supplementation to rats has been found to decrease the total bacterial cell count in the caecum( Reference Islam, Fukiya and Hagio 71 ). The microbial degradation of cholic acid to deoxycholic acid resulted in a ten times higher bactericidal activity. Bile acids also lower the gut pH and have strong antimicrobial activity, leaving only a microbiota that is tolerant to these high bile acid levels. Interestingly, cholic acid- and HFD-fed rats had the same caecal Firmicutes:Bacterioidetes ratio( Reference Islam, Fukiya and Hagio 71 , Reference Kurdi, Kawanishi and Mizutani 72 ). Bile acids activate specific nuclear receptors (farnesoid X receptor, preganane X receptor and vitamin D receptor), G protein-coupled receptor, and cell signalling pathways (c-jun N-terminal kinase 1/2, AKT and ERK 1/2) in the liver and gastrointestinal tract. Activation of nuclear receptors and cell signalling pathways alter the expression of genes encoding enzymes/proteins involved in the regulation and synthesis of bile acids( Reference Hylemon, Zhou and Pandak 73 ).
Thus, it is possible that the altered microbiota, as a consequence of HFD and saturated fats, activates the farnesoid X receptor and subsequently increases bile production. The bile acids may selectively decrease the growth of bacteria, which protect against obesity and its metabolic effects. Likewise, bile acids may promote the growth of bacteria, which increase the gut permeability and facilitate a translocation of LPS to the circulation. Several studies have shown that metabolic endotoxaemia could be generated by gut LPS absorption during digestion of a HFD( Reference Cani, Amar and Iglesias 67 , Reference Laugerette, Vors and Geloen 74 – Reference Amar, Burcelin and Ruidavets 77 ). However, none of these studies has investigated the endotoxin metabolism or inflammation in plasma and AT of different dietary fats. A recent study in mice showed that compared with milk fat, sunflower-seed oil and palm oil resulted in higher systemic and AT inflammation, whereas rapeseed oil resulted in lower inflammation( Reference Laugerette, Furet and Debard 78 ). Another study showed that the addition of both lard and fish oil to the diet promotes an interaction with the gut microbiota, which induces inflammation in AT by increasing the circulating levels of microbial products in the blood( Reference Caesar, Tremaroli and Kovatcheva-Datchary 18 ). The mice, which were fed lard for 11 weeks, had higher toll-like receptor (TLR) activation and AT inflammation and lower insulin sensitivity as compared with mice that were fed with fish oil. Additionally, knockout mice for TLR signalling were protected against lard-induced AT inflammation( Reference Caesar, Tremaroli and Kovatcheva-Datchary 18 ). Lastly, the study showed that serum levels of LPS were higher in mice fed lard compared with mice fed fish oil, indicating that lard mediates an increased uptake of LPS that may directly affect AT inflammation( Reference Caesar, Tremaroli and Kovatcheva-Datchary 18 ).
Lipopolysaccharide in the circulation and trafficking to adipose tissue
In the circulation, LPS is primarily (about 90 %) bound to lipoproteins( Reference Kallio, Buhlin and Jauhiainen 79 ). It has been shown that all plasma lipoprotein subclasses sequester LPS under simulated physiological conditions. HDL has the highest binding capacity for LPS, although HDL-bound LPS is redistributed between different classes of lipoproteins( Reference Levels, Abraham and van den Ende 80 ). Free LPS in plasma is quickly attached to soluble CD14 (sCD14) or LPS-binding protein (LBP), which are located exclusively on HDL( Reference Wurfel, Kunitake and Lichenstein 81 ). Studies on overweight and obese individuals have shown a link between poor habitual diet and low plasma levels of sCD14 levels( Reference Kong, Holmes and Cotillard 82 ). LBP catalyses a transfer of LPS to lipoproteins, transfer of LPS from LPS micelles to sCD14, and from LPS–sCD14 complexes to HDL and possibly also to other lipoproteins( Reference Wurfel and Wright 83 ). Overfeeding in healthy men has been shown to increase plasma LPS levels and decrease the LBP:sCD14 ratio( Reference Laugerette, Alligier and Bastard 84 ), suggesting a preferential sequestering of LPS in lipoproteins that may facilitate a transport of LPS to AT. Interestingly, one study has shown that intake of milk fat, compared with rapeseed oil, sunflower-seed oil and palm oil, resulted in a higher LBP:sCD14 ratio. In contrast, intake of rapeseed oil was associated with reduced inflammation, through higher levels of sCD14, despite a more pronounced endotoxaemia( Reference Laugerette, Furet and Debard 78 ). The authors proposed that HFD-induced inflammation in mice depends on the dietary fat composition. The inflammatory reaction could be partly mediated by plasma LPS receptors and transporters (sCD14 and LBP) directed toward TLR4 and CD14 activation, whereas an increased gut-derived plasma LPS concentration is a less likely initiator of inflammation( Reference Laugerette, Furet and Debard 78 ).
There is structural homology between phospholipids and LPS. Indeed, LPS is classified as a phospholipid with amphipathic properties. The phospholipid transport protein (PLTP), which regulates the transport of phospholipids between cell membranes and HDL, also transfers LPS between different classes of lipoproteins. PLTP has been shown to transfer LPS from artificial vesicles to reconstituted HDL in a model system( Reference Hailman, Albers and Wolfbauer 85 ). The rapid sequestration of LPS appears to be an innate immune response against sustained activation of cellular immunity by LPS in the host( Reference Levels, Marquart and Abraham 86 ). The evolution of this system to neutralise LPS by transfer to lipoproteins suggests that there is likewise a sophisticated system to handle the LPS-containing lipoproteins.
LPS activates pathogen-associated molecular pattern (PAMP) responses by acting as an agonist on TLR4, which increases the expression and secretion of inflammatory cytokines( Reference Medzhitov and Horng 65 ). TLR4 does not bind directly to LPS, but the activation of TLR leads to the assembly of a complex of LPS-binding proteins such as LBP, CD14 and myeloid differentiation factor-2 (MD-2). This complex recognises a common ‘pattern’ in structurally diverse LPS molecules, which results in the formation of the activated (TLR4–MD-2–LPS)2 complex( Reference Park, Song and Kim 87 ). LPS binds to CD14 and this complex functions as a fundamental chaperone and assists in the formation of the signalling complex (TLR4–MD-2–LPS)2. This complex initiates the myeloid differentiation primary response protein 88 (MyD88)-dependent pathway leading to NF-κB activation( Reference Triantafilou, Triantafilou and Fernandez 88 ). The involvement of MyD88 in HFD-induced endotoxaemia has been documented through studies where targeted reduction of MyD88 expression in intestinal cells was associated with unaltered serum levels of LPS and IL-6( Reference Everard, Geurts and Caesar 89 ). It has also been shown that CD36 knockout mice on a HFD displayed reduced AT inflammation, and adipocytes and macrophages from these mice had reduced pro-inflammatory cytokine response when exposed ex vivo to LPS( Reference Cai, Wang and Ji 90 ). Additionally, CD14 is also required for LPS-induced TLR4 endocytosis and relocalisation of the entire LPS receptor complex( Reference Zanoni, Ostuni and Marek 91 ).
It has been estimated that the human gut contains about 1 g of LPS( Reference Erridge, Attina and Spickett 75 ). However, it should be emphasised that LPS from different Gram-negative bacteria displays differences in the ability to mount an inflammatory response in cultured cells under standardised conditions( Reference Hansen, Poulsen and Wurtz 92 ). It can be speculated that both the pool size and composition of Gram-negative bacteria in the gut are important determinants for the systemic availability of LPS. LPS has been detected in the blood and plasma of healthy animals and humans at low concentrations (between 1 and 200 pg/ml)( Reference Laugerette, Vors and Geloen 74 – Reference Ghoshal, Witta and Zhong 76 ), suggesting that small amounts of LPS are constantly passing through the intestinal epithelial barrier and that the gut is a repository of immunological triggers. These concentrations are similar to the plasma concentrations (30–60 pg/ml) in human volunteers after the administration of 2–4 ng LPS/kg body weight( Reference Marshall 93 ). Therefore, if LPS is translocated to the circulation, inactivation of LPS and LPS clearance from the circulation are tremendously important in order to maintain the highly sensitive reactions of the innate immune system to the bacterial components. It has been a common notion that the intestinal mucosa is an effective barrier against LPS in the gut in healthy animals and only in a diseased state would LPS be present in the circulation. However, we have previously suggested that a HFD increases the demand of phospholipids in enterocytes to build chylomicrons (CM). This lipid transport particle has an outer layer of phospholipids, which probably is in shortage inside the cells after a HFD( Reference Hersoug, Moller and Loft 17 ). LPS in the gut may be utilised instead of phospholipids by enterocytes to assemble CM( Reference Hersoug, Moller and Loft 17 ). Additionally, LPS also binds to the scavenger receptors SR-BI and CD36( Reference Vishnyakova, Bocharov and Baranova 94 – Reference Brundert, Heeren and Merkel 96 ), which are involved in transcytosis of lipoproteins through the capillary endothelium. Thus, there may be a facilitated transport of LPS away from the blood and to the target tissue. For instance, LPS-rich CM may be phagocytised by adipocytes and macrophages( Reference Hersoug, Moller and Loft 17 ). It has been suggested that the immune system’s threshold could be modulated by LPS to adjust inflammation and resistance to infections( Reference Moreira, Texeira and Ferreira 97 ).
Delipidation of lipoproteins in adipose tissue
The AT has a high lipoprotein lipase (LPL) activity resulting in a high delipidation rate of lipoproteins with high lipid content such as CM, HDL and VLDL. As a consequence of LPL activity, phospholipids and LPS are transferred from CM, HDL and VLDL to lipid-poor lipoproteins in the capillaries to LBP in the extracellular compartment( Reference Wurfel, Kunitake and Lichenstein 81 , Reference Gazzano-Santoro, Meszaros and Birr 98 ). The latter may be internalised by adipocytes and macrophages that express CD14 receptors. A fraction of the LPS-containing remnant CM, HDL and LDL may pass the endothelium in the capillaries and be internalised by the adipocytes and macrophages (Fig. 2). This route of clearance is attractive because some of the postprandial LPS uptake could be removed from the circulation and intermediately stored in the adipocytes or macrophages.

Fig. 2 Schematic overview of the absorption of lipids after a high-fat-diet where a fraction of the absorbed lipoproteins is transported to the adipose tissue. After ingestion of a high-fat diet, lipids are preferentially absorbed in the small intestine. Degraded nutrients such as monosaccharides, amino acids and SCFA enter the bloodstream through the small venules in the villi and are transported to the liver through the hepatic portal vein. Lipopolysaccharide (LPS) can leak through the enterocytes, lining the gut surface, translocate by the paracellular route and be sequestered by the liver for detoxification. The enterocytes also absorb long-chain fatty acids and cholesterol and small quantities of LPS. These are incorporated in chylomicrons, which are secreted into the lymph. The chylomicrons enter the systemic circulation through the subclavian vein and are distributed to target tissue. The chylomicrons are partly delipidated by lipoprotein lipase forming chylomicron remnants. When chylomicrons shrink, phospholipids and LPS are translocated to HDL. A fraction of HDL and small chylomicron remnants with LPS are transcytosed through the capillary endothelium. Adipocytes are exposed to LPS when they internalise HDL, and small cell remnants. For a colour figure, see the online version of the paper.
Adipocyte inflammasomes and pyroptosis
The mammalian innate immune system has evolved a diverse set of microbial sensors such as the TLR, C-type lectin receptors, Rig-I-like receptors/helicases, AIM2 (absent in melanoma 2)-like receptors (ALR), and nucleotide-binding oligomerisation domain (NOD)-like receptors (NLR)( Reference Takeuchi and Akira 99 ). These receptor proteins orchestrate appropriate responses against microbes and microbial products, including LPS, in both extra-cellular and intra-cellular locations. The NLR and ALR family proteins sense molecules of microbial origin in the cytosol, and assemble a large multimeric signalling complex called the inflammasome( Reference Eldridge and Shenoy 100 ). The canonical inflammasome can be assembled by members of the NLR family or the PYHIN (pyrin and HIN (haemopoietic expression, interferon-inducibility, nuclear localisation) domain-containing protein) family member AIM2. The inflammasome recruits and proteolytically cleaves pro-caspase-1 to activated caspase-1 through interaction with the conserved caspase activation and recruitment domain (CARD) and the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC)( Reference Lu, Magupalli and Ruan 101 ), which subsequently proteolytically cleaves the cytokine precursors of IL-1β and IL-18 to initiate a pro-inflammatory and antimicrobial response( Reference Bauernfeind and Hornung 102 ). It has recently been shown that intracellular LPS activates pro-caspase-11 in mice, corresponding to caspase-4/5 in humans, by a non-canonical response and serves as an innate immune sensor by inducing an immune response independently of TLR4( Reference Hagar, Powell and Aachoui 103 , Reference Kayagaki, Wong and Stowe 104 ). In this process, NLR family pyrin domain containing-3 (NLRP3) and cytosolic pre-caspase-11 (caspase-4/5) are activated by LPS, but the details are not yet understood. An LPS-binding site within the CARD domain of caspase-11 (caspase-4/5) has been found and LPS binding results in oligomerisation and activation of the caspase( Reference Shi, Zhao and Wang 105 ). The activated caspase-11 (caspase-4/5) controls the assembly of ASC by NLRP3( Reference Eldridge and Shenoy 100 ). It has been shown that transfection of LPS into human monocytes, fibroblasts (HeLa cells) or promyelocytic leukaemia (HL60) cells promote pyroptosis, whereas attempts to find the cytosolic LPS receptor, which was suspected to contain a CARD domain, have not been successful( Reference Shi, Zhao and Wang 105 ). Nevertheless, purified recombinant caspases-4/5/11 from bacteria, but not caspases from insects, stimulated caspases to undergo auto-oligomerisation. This indicates that caspases oligomerise because of bacterial impurities such as LPS( Reference Smith, Wang and Yin 106 ). Indeed it was shown that LPS binding is required for caspase-4/5/11 oligomerisation, activation and induction of pyroptosis( Reference Shi, Zhao and Wang 105 ). Pyroptosis requires activation of caspase-4/5/11, but it is independent of caspase-1 and the adaptor protein ASC( Reference Smith, Wang and Yin 106 ). Therefore, it is possible that the detection of LPS in the cytosol activates caspase-4/5/11, which subsequently promotes the assembly of ASC via NLRP3. This activates caspase-1, leading to activation of pro-IL-1β and pro-IL-18 without pyroptosis. However, LPS-activated caspase-4/5/11 also can directly induce pyroptosis( Reference Eldridge and Shenoy 100 , Reference Smith, Wang and Yin 106 ). Interestingly, it has recently been shown that the AT in leptin-deficient and HFD-induced obese mice have hypertrophic adipocytes that express NLRP3 inflammasome activation, active caspase-1 and a high level of pyroptotic cells( Reference Giordano, Murano and Mondini 14 ). This suggests that pyroptosis of large adipocytes, mediated by activated NLPR3 inflammasomes and caspase-1, occurs as a consequence of exposure to cytosolic LPS and activation of caspase-4/5/11.
Adipocyte size, exposure to lipopolysaccharide and pyroptosis
There is an upper limit for the expansion of large hypertropic adipocytes because of factors such as hypoxia and differential matrix mechanisms( Reference Halberg, Khan and Trujillo 107 ). We hypothesise that adipocyte death size is reached when the intracellular concentration of LPS initiates pyroptosis. The intracellular concentration of LPS may therefore depend on of the metabolic activity of the cell and the amount of LPS that is translocated from lipoproteins. LPS is incorporated into CM and subsequently transported by the lymph to the blood, where it can be translocated to other lipoproteins( Reference Hersoug, Moller and Loft 17 ). Different adipocyte death sizes could reflect a difference in LPS exposure. The existence of an adipocyte death size is supported by the observation that the adipocyte cell size increases with adiposity level and it reaches a plateau in heavily obese individuals( Reference Laforest, Labrecque and Michaud 108 ). This hypothesis is supported by observations that infusion of LPS resulted in reduced adipocyte size in wild-type mice, whereas there was no effect in CD14 knockout mice( Reference Cani, Amar and Iglesias 67 ).
LPS molecules can self-aggregate to supra-molecular structures with a critical micelle concentration of 10–20 µg/ml( Reference Santos, Silva and Castanho 57 ). It has been suggested that LPS micelles activate the caspases( Reference Smith, Wang and Yin 106 ). Non-canonical activation of caspase-11 in cells exposed to intracellular LPS has been reported(103, Reference Kayagaki, Wong and Stowe 104 ). In the intracellular environment, LPS might be unevenly distributed in the cytoplasm. LPS concentrations might locally reach the critical micelle concentration and interact with caspase-4/5/11( Reference Smith, Wang and Yin 106 ). In addition, caspase activation might be mediated by the trapping of LPS in the phospholipid layer surrounding the lipid droplets. LPS can move freely in the phospholipid layer; it may self-aggregate at a critical concentration and activate caspase-4/5/11 (Fig. 3). The latter may be the most likely mechanism for the activation of caspase-4/5/11 because it has been shown that internalised LPS co-localises with membrane compartments inside cells( Reference Vishnyakova, Bocharov and Baranova 94 ). This is supported by observations showing that LPS preferentially segregates into ordered domains in giant unilamellar vesicles( Reference Henning, Sanchez and Bakas 109 ). The positive residues in the CARD domain of caspase-4/5/11 may bind to the negatively charged polar head of LPS aggregates or micelles. Caspases are recruited on the surface of these LPS structures and the close proximity of caspases on micelles stimulates oligomerisation, which is a prerequisite for activation of caspases. This can start a self-propagating signalling cascade that rapidly leads to pyroptosis( Reference Smith, Wang and Yin 106 ). Thus, we hypothesise that LPS locally reaches the critical micelle concentration and/or aggregate at the surface of lipid droplets, which in turn starts the signalling cascade, leading to pyroptotsis in the highly active and large adipocytes. In the hypertrophic AT, pyroptotic cells can recruit macrophages to remove cell remnants and debris, forming crown-like structures. It has been described that the development of crown-like structures occurs concurrently with the infiltration of neutrophils and macrophages that actively engulf remnant lipids from dead adipocytes in AT during acute lipoatrophy( Reference Murano, Rutkowski and Wang 110 ). It has been suggested that inflammation is a direct cause of pyroptosis( Reference Murano, Rutkowski and Wang 110 ). VAT is more inflamed than SAT during weight gain, suggesting that the higher rate of pyroptosis in VAT is related to a higher transfer of LPS to VAT. Thus, LPS might be a direct cause of inflammation. AT with a higher potential to expand by hyperplasia may have lower ratio of large to small adipocytes. It means that there might be fewer adipocytes undergoing pyroptosis in types of AT with a tendency to enlarge by hyperplasia if the adipocyte death size is caused by LPS. This further implies that AT, which expand primarily by hyperplasia, might be healthier than AT that expand by hypertrophy.

Fig. 3 Schematic overview of lipopolysaccharide (LPS)-induced caspase-4/5/11 activation. LPS (red-orange) binds to the caspase activation and recruitment domain (CARD) (purple) of caspase-4/5/11 (green), inducing its oligomerisation and subsequent proteolytic cleavage of pro-caspase-1 into active caspase-1 enzyme, which further cleaves proforms of the inflammatory cytokines IL-1β and IL-18 into their active forms. Possibly it is the positively charged residues on the caspase CARD domain that interact with the negatively charged LPS lipid head group. Once multiple caspase molecules are localised to micelles or to LPS aggregates in phospholipid monolayers, they may induce an auto-activation of the caspase to its active, oligomeric state. Upon activation, caspase-4/5/11 (yellow with rose corona) may cause pyroptosis or inflammation through caspase-1. ASC, apoptosis-associated speck-like protein containing a CARD; Casp1, caspase-1; NLR, nucleotide-binding oligomerisation domain (NOD)-like receptor. For a colour figure, see the online version of the paper.
Systemic and adipose tissue-specific insulin sensitivity
The accumulation of pro-inflammatory cells in VAT is an important cause of insulin resistance, which in turn is associated with type 2 diabetes. Immune cells produce cytokines and other factors that contribute to insulin resistance( Reference Lackey and Olefsky 111 ). Circumstantial experimental evidence also suggests that LPS impairs the liver’s crucial function in maintaining whole-body glucose metabolism. It has been shown in animal models of periodontitis that LPS-stimulated macrophages from the gingival sulci secrete TNF-α, IL-1β and IL-6( Reference Ilievski, Cho and Katwala 112 ). Systemic translocation of these cytokines and/or LPS from the gingival sulci may stimulate TLR4 receptors on Kupffer cells in the liver to secrete pro-inflammatory cytokines, which may result in glucose intolerance and insulin resistance( Reference Ilievski, Cho and Katwala 112 ). An altered generation of SCFA may be observed during overfeeding, obesity and the metabolic syndrome. However, studies have shown both an increased( Reference Shepherd, Ponder and Burk 113 – Reference Rahat-Rozenbloom, Fernandes and Gloor 117 ) and decreased( Reference Murphy, Cotter and Healy 118 , Reference Murugesan, Ulloa-Martinez and Martinez-Rojano 119 ) production of SCFA during overfeeding, obesity and the metabolic syndrome. Recently, it was shown that an increased production of acetate, due to a gut microbiota–nutrient interaction in HFD-fed rodents, leads to activation of the parasympathetic nervous system. This resulted in increased secretion of ghrelin and augmented glucose-stimulated insulin secretion (GSIS). This generates a positive feedback loop, resulting in hyperphagia, hypertriacylglycerolaemia, ectopic lipid deposition in the liver and skeletal muscle, and liver and muscle insulin resistance( Reference Perry, Peng and Barry 120 ). In order to investigate a causal relationship between microbiota and GSIS, faecal material was transferred from chow- or HFD-fed donor rats to chow- or HFD-fed recipients. It was shown that the Firmicutes:Bacteroidetes ratio was higher in fresh faecal pellets from HFD-fed donors compared with chow-fed donors. In addition, the faecal transplantation changed the microbiome, acetate turnover, faecal acetate and GSIS in the recipient animal to resemble that of the donors( Reference Perry, Peng and Barry 120 ). The effect of acetate on GSIS was mediated by a central parasympathetic outflow. It has been proposed that both the sympathetic and the parasympathetic arm of the autonomic nervous system innervate AT( Reference Kreier and Buijs 121 ). Sympathetic activity induces catabolic effects (lipolysis), whereas parasympathetic activity induces anabolic effects (lipogenesis)( Reference Kreier and Buijs 121 , Reference Kreier, Fliers and Voshol 122 ). However, it has not been possible to verify the existence of parasympathetic innervations in AT( Reference Bartness, Liu and Shrestha 123 ). Insulin resistance in AT is governed by the local secretion of cytokines. The expressions of NLRP3 inflammasome and caspase-1 by macrophages and adipocytes are associated with the severity of obesity, insulin resistance and type 2 diabetes( Reference Vandanmagsar, Youm and Ravussin 124 ). This is mediated by the elevated production of IL-1β and IL-18. These cytokines may reduce the secretion of insulin and decrease glucose uptake, which are hallmarks of insulin resistance( Reference Vandanmagsar, Youm and Ravussin 124 ).
Conclusion
Obesity is characterised by low-grade inflammation, which may originate from AT, although the underlying molecular mechanisms are largely unknown. A moderate increase in the plasma concentration of LPS, translocated from the gut, may be implicated in metabolic diseases. We have outlined a coherent chain of events that links postprandial lipid absorption, composition of the intestinal microbiome, endotoxaemia and metabolic disease. The evidence suggests that LPS is directly and indirectly involved in the histological and biological changes that occur in AT during obesity. The hypotheses can be summarised as follows: (1) LPS is involved in defining the adipocyte death size and formation of crown-like structures; (2) LPS is involved in the transition of macrophages with the M2 phenotype to the M1 phenotype; (3) LPS is responsible for the activation of caspase-4/5/11 and the induction of pyroptosis in adipocytes; and (4) LPS oligomerises in lipid droplet membranes and subsequently activates caspase-4/5/11. Further studies are warranted to test the hypotheses and subsequently integrate the information into a coherent description of the effects of HFD and microbiota-derived LPS as a mechanism of action of obesity.
Acknowledgements
L.-G. H. conceived and prepared the initial draft. P. M. and S. T. critically reviewed the draft. All authors read and approved the final draft of the manuscript.
There are no conflicts of interest.



