
Volume 57 - Issue 3 - June 2009
Contents
Article
Selectivity of Co and Ni by K-depleted micas
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 279-289
-
- Article
-
- You have access
- Export citation
Simple synthesis and characterization of nanoporous materials from talc
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 290-301
-
- Article
-
- You have access
- Export citation
Application of 29Si and 27Al MAS NMR spectroscopy to the study of the reaction mechanism of kaolinite to illite/muscovite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 302-310
-
- Article
-
- You have access
- Export citation
The occurrence and origin of the söğüt kaolinite deposits in the Paleozoic Saricakaya granite-granodiorite complexes and overlying Neogene sediments (Bilecik, northwestern Turkey)
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 311-329
-
- Article
-
- You have access
- Export citation
Synthesis of a Mg-Cd-Al layered double hydroxide and sorption of selenium
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 330-337
-
- Article
-
- You have access
- Export citation
Three new, quick CEC methods for determining the amounts of exchangeable calcium cations in calcareous clays
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 338-352
-
- Article
-
- You have access
- Export citation
Factors governing the formation of lithiophorite at atmospheric pressure
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 353-360
-
- Article
-
- You have access
- Export citation
-
Lithiophorite is a naturally occurring Mn oxide mineral commonly found in soils and sediments. The usual method of synthesizing lithiophorite is via a hydrothermal process in an autoclave at relatively high temperature and pressure. In the present study, an alternative, reflux method, at atmospheric pressure, for synthesis of lithiophorite was developed successfully. The influence of reaction duration, temperature, type of precursor birnessite (H-birnessite, Na-birnessite, aged Na-birnessite), and pH on the formation of lithiophorite were investigated by reflux treatment of lithium-aluminum hydroxide complex ion (
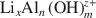 )-exchanged birnessite. The results show that the degree of conversion of lithiophorite decreases with decreasing reaction temperature. Lithiophorite can be obtained at pH values from 5.0 to 9.0, but a circumneutral pH is more favorable for formation at atmospheric pressure. Conversion of Na-birnessite (Bir-OH) to lithiophorite is more favored than aged Na-birnessite (Bir-OH-A). Lithiophorite was not obtained by refluxing the
)-exchanged birnessite. The results show that the degree of conversion of lithiophorite decreases with decreasing reaction temperature. Lithiophorite can be obtained at pH values from 5.0 to 9.0, but a circumneutral pH is more favorable for formation at atmospheric pressure. Conversion of Na-birnessite (Bir-OH) to lithiophorite is more favored than aged Na-birnessite (Bir-OH-A). Lithiophorite was not obtained by refluxing the 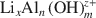 ion-exchanged H-birnessite (Bir-H) sample. The rate of conversion of lithiophorite increases with increasing reflux time. Lithiophorite synthesized by a reflux process has pseudo-hexagonal crystals of 0.1–0.5 µm with a chemical composition of Li0.24Al0.46MnO2.67(H2O)1.25. The results have important implications for the origin and underlying mechanism of lithiophorite formation in the environment.
ion-exchanged H-birnessite (Bir-H) sample. The rate of conversion of lithiophorite increases with increasing reflux time. Lithiophorite synthesized by a reflux process has pseudo-hexagonal crystals of 0.1–0.5 µm with a chemical composition of Li0.24Al0.46MnO2.67(H2O)1.25. The results have important implications for the origin and underlying mechanism of lithiophorite formation in the environment.
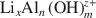 )-exchanged birnessite. The results show that the degree of conversion of lithiophorite decreases with decreasing reaction temperature. Lithiophorite can be obtained at pH values from 5.0 to 9.0, but a circumneutral pH is more favorable for formation at atmospheric pressure. Conversion of Na-birnessite (Bir-OH) to lithiophorite is more favored than aged Na-birnessite (Bir-OH-A). Lithiophorite was not obtained by refluxing the
)-exchanged birnessite. The results show that the degree of conversion of lithiophorite decreases with decreasing reaction temperature. Lithiophorite can be obtained at pH values from 5.0 to 9.0, but a circumneutral pH is more favorable for formation at atmospheric pressure. Conversion of Na-birnessite (Bir-OH) to lithiophorite is more favored than aged Na-birnessite (Bir-OH-A). Lithiophorite was not obtained by refluxing the 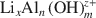 ion-exchanged H-birnessite (Bir-H) sample. The rate of conversion of lithiophorite increases with increasing reflux time. Lithiophorite synthesized by a reflux process has pseudo-hexagonal crystals of 0.1–0.5 µm with a chemical composition of Li
ion-exchanged H-birnessite (Bir-H) sample. The rate of conversion of lithiophorite increases with increasing reflux time. Lithiophorite synthesized by a reflux process has pseudo-hexagonal crystals of 0.1–0.5 µm with a chemical composition of LiSurface properties of illite-smectite minerals as detected by interactions with rhodamine 6G dye
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 361-370
-
- Article
-
- You have access
- Export citation
Application of chemical geothermometry to low-temperature trioctahedral chlorites
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 371-382
-
- Article
-
- You have access
- Export citation
-
Low-temperature chlorites formed in diagenetic to low-grade metamorphic environments generally have greater Si contents and larger numbers of octahedral vacancies, and smaller Fe+Mg contents than higher-grade metamorphic chlorites. The compositional variations are characterized approximately by four end-member components: Al-free trioctahedral chlorite, chamosite, corundophilite, and sudoite. The solid solution is considered to be a random mix of cations and vacancies in the octahedral sites. Using the compositions of chlorites from Niger, Rouez, and Saint Martin diagenetic-hydrothermal series, a new, more convenient geothermometer, applicable to low-T chlorites is proposed and comparison made with geothermometers proposed previously. The chlorites studied contain appreciable amounts of Fe(III) (>14% of the total Fe), determined by Mössbauer spectroscopy. The calculations under which all Fe was regarded as ferrous gave considerable overestimates for the formation temperature, irrespective of the geothermometer used. This problem was reduced by taking into account the presence of Fe(III) in the octahedral sites. The geothermometer from this study gave more reasonable estimates than the geothermometers proposed by Walshe (1986) and Vidal et al. (2001), particularly in the case of the Niger chlorites which crystallized in the lowest-temperature conditions. The ordered-site substitution model of solid solution developed by Vidal et al. (2001) predicted satisfactorily the formation temperature of the Rouez chlorites and of some of the Saint Martin chlorites, suggesting that the chlorite compositions are controlled by the
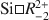 exchange at low-T conditions while they are controlled by Tschermak exchange at higher temperatures. The decreasing number of vacancies with temperature are poorer in Fe-rich than in Fe-poor chlorites. Furthermore, the ordered-site occupation of cations and vacancies in trioctahedral chlorite occurs concomitantly with the compositional changes ruled by increasing temperature conditions.
exchange at low-T conditions while they are controlled by Tschermak exchange at higher temperatures. The decreasing number of vacancies with temperature are poorer in Fe-rich than in Fe-poor chlorites. Furthermore, the ordered-site occupation of cations and vacancies in trioctahedral chlorite occurs concomitantly with the compositional changes ruled by increasing temperature conditions.
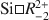 exchange at low-
exchange at low-Isomerization of 1-butene catalyzed by surfactant-modified, Al2O3-pillared clays
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 383-391
-
- Article
-
- You have access
- Export citation
Near-infrared spectroscopic analysis of acid-treated organo-clays
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 392-403
-
- Article
-
- You have access
- Export citation