


Introduction
Codes of ethics and regulations have been created to guide researchers worldwide in conducting research with human subjects. In the United States, the Belmont Report and the Common Rule guide ethical research conduct. The Common Rule, officially called “Basic Department of Health and Human Services (HHS) Policy for the Protection of Human Research Subjects,” mandates that research participants are given detailed information on the purpose of the study so that they can make informed, autonomous decisions before agreeing to participate [1]. This informed consent process ensures that research participants are properly informed of all aspects of the study. Obtaining informed consent is considered responsible conduct in human subjects research [Reference Tamariz2].
Under the Common Rule, researchers use informed consent forms to document and provide specific details regarding a research study, including risks, benefits, and requirements for study participation [Reference Larson, Foe and Lally3]. The Office for Human Research Protections recommends that written consent forms contain easy-to-read and understandable information so that the lay person can make an informed decision regarding participation in research studies [4]. However, this is difficult to accomplish because the Common Rule mandates up to 18 elements that must be disclosed to potential research participants—9 basic elements and 9 research dependent elements [1]. Because of this, most written consent forms are lengthy and difficult to read for research participants, particularly those with low health literacy [Reference Larson, Foe and Lally3, Reference Manta5–Reference Sherlock and Brownie7].
Consent forms used in research studies are known to be more difficult to understand than other informed consent documents because of the increased disclosure requirements, which is often too much information for the average reader to process at once [Reference Tamariz2, Reference Manta5, 8]. In addition, research-related consent forms typically include complex information regarding study procedures, contain several unfamiliar medical and research terms, and are often required to include difficult to understand legal terminology [Reference Tamariz2, Reference Montalvo and Larson9]. As a result, it is very common for research participants to have difficulty understanding the information written in the consent form before agreeing to participate in the study, leaving many to wonder if participants are actually providing informed consent [Reference Montalvo and Larson9–Reference Sugarman and Paasche-Orlow12]. Although it is recommended that consent forms are written at a sixth to eighth grade reading level or lower [Reference Tamariz2, Reference Hochhauser13, Reference Denzen14], most research-related consent forms are written at a level much higher than participants’ reading skill levels [8, Reference Donovan-Kicken15, Reference Paasche-Orlow16]. A survey by the National Assessment of Adult Literacy reported that the average reading level of US adults is eighth grade or below [Reference Kutner17]. A recent review of written consent forms revealed that most are currently written at a 10th grade reading level or higher across all medical specialties [Reference Larson, Foe and Lally3].
Consent documents that are written at a high grade level may create additional risks for research participants due to lack of understanding [Reference Nishimura18]. To ensure that participants are fully informed of all aspects of the research project and completely understand what is expected when agreeing to participate, it is important to make sure that consent forms are written in plain language, that is, writing that is clear and easy to understand the first time the participant reads or hears it [19]. To that end, the newly enacted Final Rule for the Protection of Human Subjects (the revised Common Rule), which goes into effect in January 2018, still requires that the consent be “in language understandable to the subject” and mandates that “the informed consent must be organized in such a way that facilitates comprehension [20].”
The new Final Rule will benefit participants because consent forms that are concise and written in plain language have many benefits. First, plain language consent forms are easy to read and understand [Reference Kim and Kim21] and may lead to increased participation from those in vulnerable populations [Reference Nishimura18]. Second, plain language consent forms help research participants to make informed decisions regarding participation; after all, the purpose of the consent form is to obtain participant consent that is fully and freely given after being informed of the relevant study information. In addition, consent forms that are easy to understand should reduce the burden on Institutional Review Board (IRB) members who review and approve all consent forms prior to implementation in studies.
Effective health literacy approaches can improve the readability of consent forms. One recommended health literacy approach is to simplify the written content by eliminating jargon and using words that are short and easy to understand [Reference Tamariz2, Reference Kim and Kim21–Reference Stunkel23]. Consent forms should be formatted in a way that allows for easy reading, including use of adequate white space, large fonts, and bolding of text where needed [Reference Kim and Kim21, Reference Kass24]. While there is limited research linking improved reading level and comprehension, simplifying consent forms using plain language editing techniques and adhering to the recommended reading grade level (sixth grade or below) makes the document easier to read and understand [Reference Tamariz2, Reference Nishimura18, Reference Kim and Kim21, Reference Kass24]. The purpose of this research study was to assess the readability of IRB approved informed consent documents, to implement an intervention to improve the readability of IRB approved consent forms, and to measure the impact of that intervention over 1 year at an academic research institution.
Materials and Methods
To establish a baseline before intervention, we conducted a retrospective analysis of 217 IRB approved, investigator initiated written informed consents from 2013 to 2015 to determine readability. The mean grade level readabilities were derived from the results of 3 readability formulas (Fry Graph [Reference Fry25], Flesch-Kincaid [Reference Flesch26], and Simple Measure of Gobbledygook (SMOG) Index [Reference McLaughlin27]) using online readability tools. Results were validated by expert hand calculations for random samples of the set of informed consents evaluated.
The intervention consisted of developing an informed consent template adhering to plain language and health literacy best practices for written communication [28–30] that meets IRB and regulatory requirements. This template was developed through an iterative process with research participant input and collaboration among the University of Arkansas for Medical Sciences (UAMS) Center for Health Literacy, UAMS IRB, a research ethicist, and the UAMS Translational Research Institute, the home of the institution’s Clinical and Translational Science Award (CTSA). Each iteration of template content was assessed for readability by the Center for Health Literacy to ensure that each time an element was added or changed, the template adhered to plain language best practices. When all contributors were satisfied with the content, after over a year of development, the template was presented in a focus group of patients with low health literacy. Methods of soliciting qualitative information and overcoming barriers to meeting low health literacy needs were employed [Reference Hadden31] throughout the focus group. The group was facilitated by trained professionals at the Center for Health Literacy.
The plain language informed consent template was made available on the institution’s IRB Web site and promoted in internal communications to investigators. Additionally, brief trainings were provided to IRB committees on the availability and utility of the template in regularly scheduled meetings. All IRB committees were provided with the training over a 4-month period.
The readability level of the template is the fifth grade. The template was made available to investigators as an optional resource on the IRB Web site, and IRB committees were trained on how to best use the template for approval purposes. Additionally, investigators were provided with examples of content in plain language to add for the customized portions of the form. The same readability assessment methods were employed on the prospective sample of IRB approved informed consents for 1 year following implementation of the intervention.
Results
The retrospective analysis revealed a mean readability of 10th grade for written informed consents from 2013 to 2015 (n=217) (see Fig. 1). Post-intervention results show a shift in the mean readability to the eighth grade level (see Fig. 2). A 49% adoption rate of the template (n=41) resulted in written informed consents with a mean readability of seventh grade (range 6th–10th grade, 90.2% ≤8th grade), compared with a mean of 10th grade (range 7th–12th grade, 11.9% ≤8th grade) for the no adoption group (n=42). The intervention has resulted in a 658% increase in the percentage of written informed consents at eighth grade readability or below compared with baseline. The effect of the plain language template on readability was substantial; 90% of the informed consents that were in the template were assessed to be in the recommended readability range and 88% of the informed consents that were not in the template were assessed to be above the recommended range (see Fig. 3).
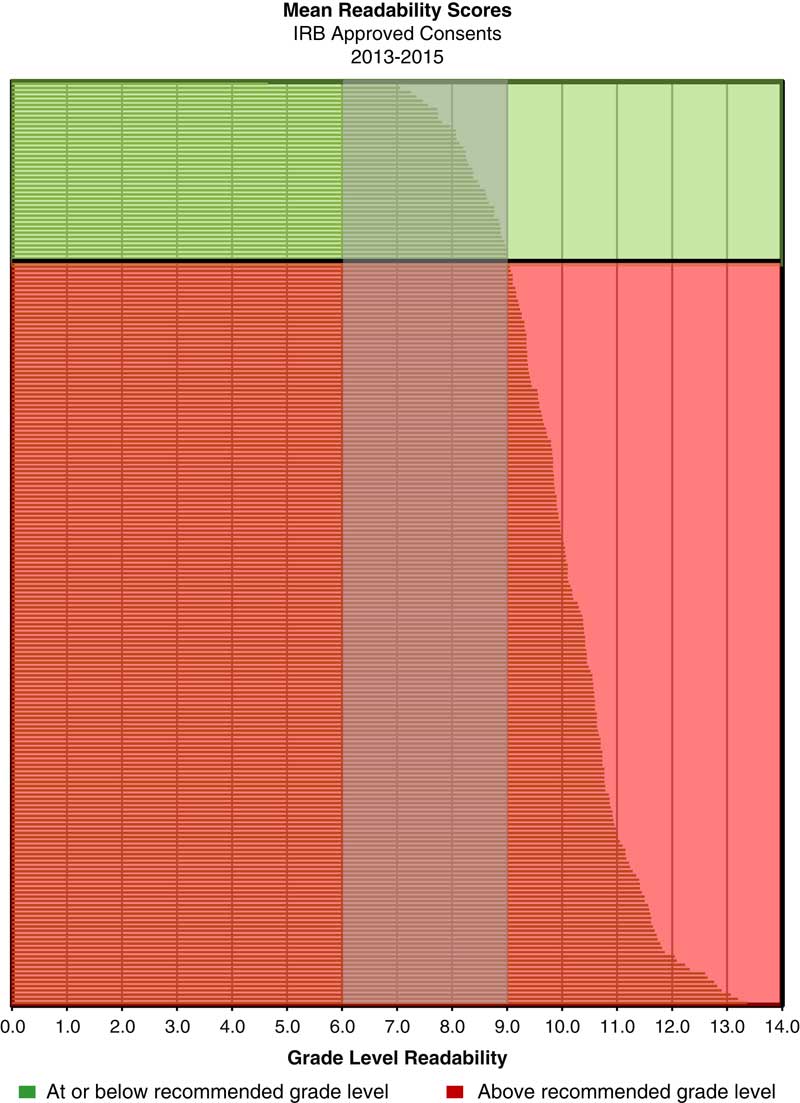
Fig. 1 Mean readability scores for Institutional Review Board (IRB)-approved consents: 2013–2015 (n=217)
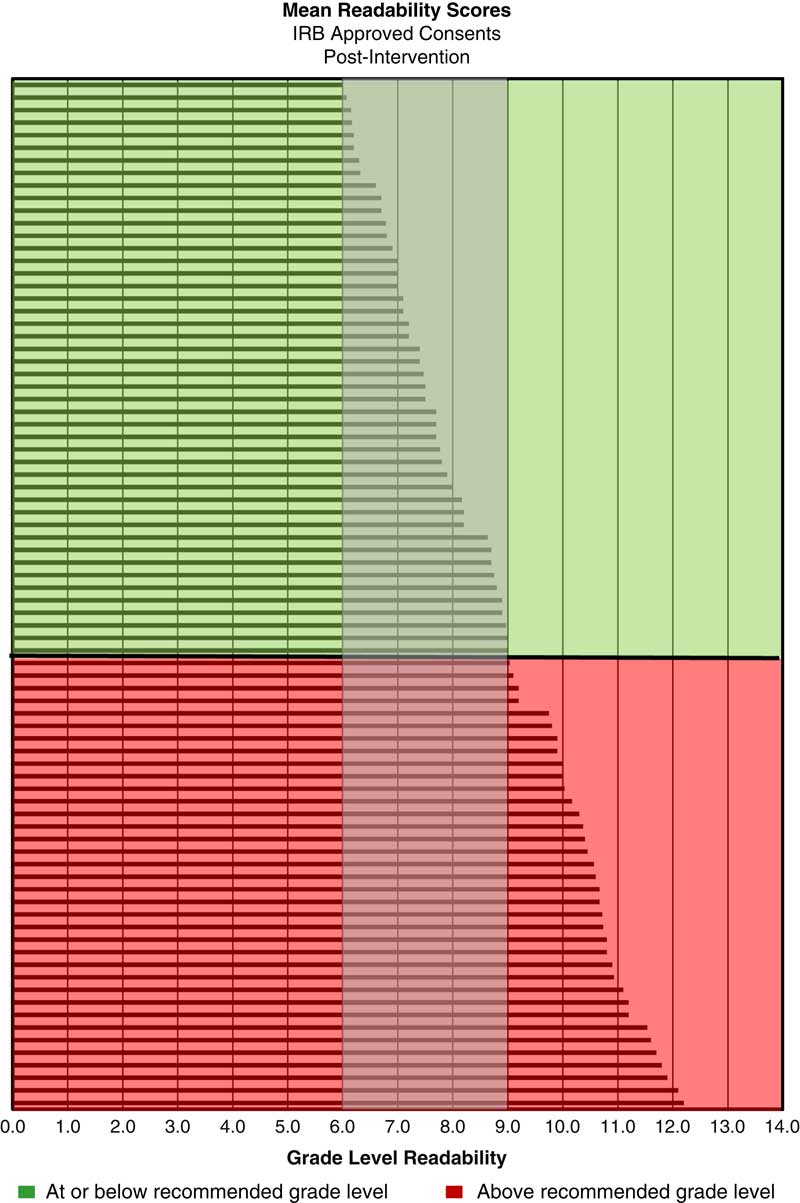
Fig. 2 Mean readability scores for Institutional Review Board (IRB)-approved consents post-intervention (n=82)
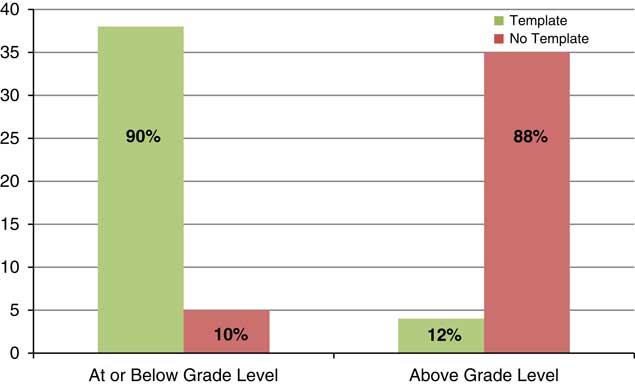
Fig. 3 Institutional Review Board-approved consents within grade-level recommendations post-intervention
Conclusions
Low health literacy is common in individuals who experience healthcare disparities and can limit participation in clinical research. Few studies have examined interventions to address this barrier to research. The present study demonstrates feasibility for using a clear, understandable informed consent template in investigator initiated research. Our results show a 49% template adoption rate over the last year. Moreover, this study demonstrates not only the success of our intervention, but the importance of stakeholder engagement among CTSA leadership, health literacy experts, the IRB, investigators, and research participants in the development and testing of an intervention to develop written informed consents “understandable to the subject” while adhering to all regulatory requirements.
There are limitations of this study. First, we only assessed written informed consents from investigator initiated research because we wanted to determine if providing investigators with plain language tools and trainings would change their behaviors in obtaining consent and when submitting forms to our IRB. Industry sponsored, multi-site consortium, and cancer trials are often overseen by external IRBs, and as such, investigators may have little control over changes to them. We hope that our results will drive changes for all types of research consent, regardless of the source of the form. The new Common Rule supports these changes. Lastly, we know that readability does not, in and of itself, assess participant understanding. Our future research will use mixed methods to evaluate and understand how well our health literate template meets the needs and preferences of research participants. We will aim to assess both comprehension of risks and benefits as well as decisional confidence in future studies.
Acknowledgments
The authors thank other contributors from UAMS to this research: Wendy Thompson, Dr. Alison Oliveto, Nancy Dockter, Leah Eisenberg, and Edith Paal. The project described was supported by the Translational Research Institute (TRI), grant UL1TR000039 through the NIH National Center for Research Resources and National Center for Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosures
The authors have no conflicts of interest to declare.




 Open access
Open access