

Introduction
Genetic Diagnosis of Early Infantile Encephalopathy (EIEE)
EIEE is a rare but debilitating neurological disorder characterized by frequent, typically intractable seizures within the first months of life, severe psychomotor deficits, and low survival rate beyond infancy. Multiple causes for EIEE are known, including genetic mutations, metabolic disorders, and structural abnormalities of the brain. Genetic cases of EIEE are often caused by dominant de novo mutations in voltage-gated ion channel genes required for neuronal development, including SCN1A, SCN2A, SCN8A, and KCNQ2 [Reference de Kovel1]. When a genetic cause for EIEE is suspected, known causative mutations may be detected using commercially available gene panel sequencing tests. However, panel tests yield a genetic diagnosis in only 10%–50% of cases [Reference Carvill2–Reference Kodera4], possibly because there are over 300 genes [Reference de Kovel1] known to cause EIEE, and different panels include only subsets of these genes. Furthermore, EIEE disease presentation may mimic other seizure disorders, and multiple rounds of testing may be required to eliminate conditions such as Angelman Syndrome before EIEE is confirmed.
When genetic tests fail to yield a diagnosis for EIEE, sequencing and analysis of whole exomes or genomes may detect disease-causing variants. However, analysis of genomic sequences is more expensive than panel testing, and requires either familiarity with command-line software tools and significant computational resources, or collaboration with a bioinformatics expert or commercial service provider. Such analyses are costly and time-consuming, limiting the adoption of whole exome or genome-based approaches for routine clinical diagnoses.
The Iobio Tool Suite Facilitates Rapid and Intuitive Genome Analysis
We have developed iobio, a web-based genome sequence analysis system (http://iobio.io). Iobio is designed to enable nonexpert clinicians and researchers to analyze genome sequence data and obtain genetic diagnoses, using standard equipment such as a laptop or desktop computer and a web browser. Iobio uses an intuitive user interface and a series of web applications to perform discrete genome sequence analyses in an interactive, visually driven fashion. Here we demonstrate the use of the iobio tool suite to successfully analyze the whole genome sequence of a female infant with EIEE for whom initial gene panel testing was nondiagnostic.
Methods
Sequencing and Data Acquisition
Patients were recruited locally through the Pediatric Neurology Clinic at Primary Children’s Hospital. This study was approved by the University of Utah Institutional Review Board. Medical history and electroencephalogram findings were reviewed to confirm the diagnosis of EIEE. Magnetic resonance imaging (MRI) and laboratory data were reviewed to confirm that patients did not have other causes of EIEE. Exclusion criteria included the presence of an inborn error of metabolism, an established diagnosis of a genetic syndrome, or structural brain abnormality. Proband and both parents were enrolled in the study and provided blood samples. DNA was extracted from the blood, and whole genome sequencing (WGS) was performed on Illumina 10×sequencers, at 60×nominal genome coverage for each sample. The sequencing reads were mapped with a “best practices” mapping pipeline using the BWA software [Reference Li and Durbin5]. The resulting alignment (BAM [Reference Li6]) files were stored locally and accessed using the iobio software tools in the Google Chrome internet browser.
Quality Control
BAM files were accessed using the bam.iobio [Reference Miller7] sequence alignment inspector web app (http://bam.iobio.io) from the University of Utah data laboratory information management system GNomEx [Reference Nix8], where links for all BAM files are provided. Quality metrics, including read coverage distribution, percentage of mapped reads, and duplication rate were inspected visually in the web browser using the graphics returned by the software.
Variant Calling and Prioritization
BAM files were accessed from within the gene.iobio web app (http://gene.iobio.io) by clicking the “Files” button, selecting the relevant samples, and then clicking “Load.” Variant calling was performed on the selected BAM files within gene.iobio using the integrated FreeBayes [Reference Garrison and Marth9] calling algorithm. A prioritized list of the top 20 genes associated with EIEE was obtained within gene.iobio by clicking “Genes” and entering the key phrase “early infantile epileptic encephalopathy” in the “Phenolyzer” field to generate a list of genes most likely associated with one or a combination of input phenotype terms, using the Phenolyzer [Reference Yang, Robinson and Wang10] tool. All variants in this gene list were assessed by clicking the “Analyze all genes” button within gene.iobio. Identification of genes harboring candidate variants was achieved by clicking the “Filter” button and applying successive levels of filtering. The “Known Pathogenic” button was selected to highlight any genes containing rare variants (<1% allele frequency in the thousands of samples present in the 1000 Genomes Project [11, 12] and ExAC [Reference Lek13] databases) that are already identified as pathogenic, or likely pathogenic in ClinVar [Reference Landrum14, Reference Landrum15]. To search for genes containing rare de novo variants that are predicted to have a “High” or “Moderate” functional impact, the “De novo VUS” button was selected (variants of unknown significance). Returned genes were then individually selected to give a visual representation of the variants present that conform to the de novo filter, along with data coverage across the gene in the context of the gene model. Variants with a severe predicted functional effect were further investigated using links within gene.iobio to determine their significance and likelihood of disease causation.
Results
The subject of our study is a female infant who presented with a confusing mixture of seizures, hypotonia, and apneic episodes in the first week of life. The seizures persisted, and then the patient also developed severe neurodevelopmental impairment. Because of the mixture of hypotonia and epilepsy, a wide range of diagnostic tests were ordered, including brain MRI, testing for inborn errors of metabolism, testing for causes of hypotonia (including for Prader-Willi Syndrome and for muscular dystrophies), and testing for epilepsy disorders with specific treatment implications (such as pyridoxine-dependent epilepsy and glucose transporter defect). Genetic testing (all of which was normal) included a SNP microarray, and a gene panel for causes of epilepsy and neurodevelopmental impairment (which included ARX, ATRX, CDKL5, FOXG1, MEF2C, MED17, NRXN1, OPHN1, PCDH19, PNKP, SLC2A1, SLC9A6, TCF4, UBE3A, ZEB2). The patient was subsequently enrolled in the present study, in which WGS was carried out to identify potentially causative de novo mutations. The WGS data was processed with a standard read mapping pipeline at the USTAR Center for Genetic Discovery. When alignment (BAM) files were available, we used the iobio tools to confirm data quality and to identify the likely causative de novo variant causing EIEE in the patient.
To ensure reliable diagnoses, an evaluation of the quality of the underlying data prior to variant analysis was performed. We investigated data quality parameters using bam.iobio for all 3 samples in the EIEE trio. Data quality statistics were generated in <10 seconds per sample. Fig. 1 reports the sequence alignment metrics for the proband: corresponding metrics for the 2 parents were similar. The read coverage showed a Poisson distribution centered on ~80×, higher than the expected value of 60×. Sequencing fragment length (averaging roughly 325 bp), mapping rate (>99% of reads mapped), and mapping quality (a large fraction of the reads mapping with mapping quality (MQ) >40) were as expected for high quality sequence data. However, this sequencing library had unusually large (16.9%) polymerase chain reaction (PCR) fragment duplication rate (the optimal rate is <5%). Although the high duplication rate meant that overall effective sequence coverage was only about 5/6th of the measured >80×, the resulting >60× effective coverage was still sufficient for accurate de novo mutation detection.
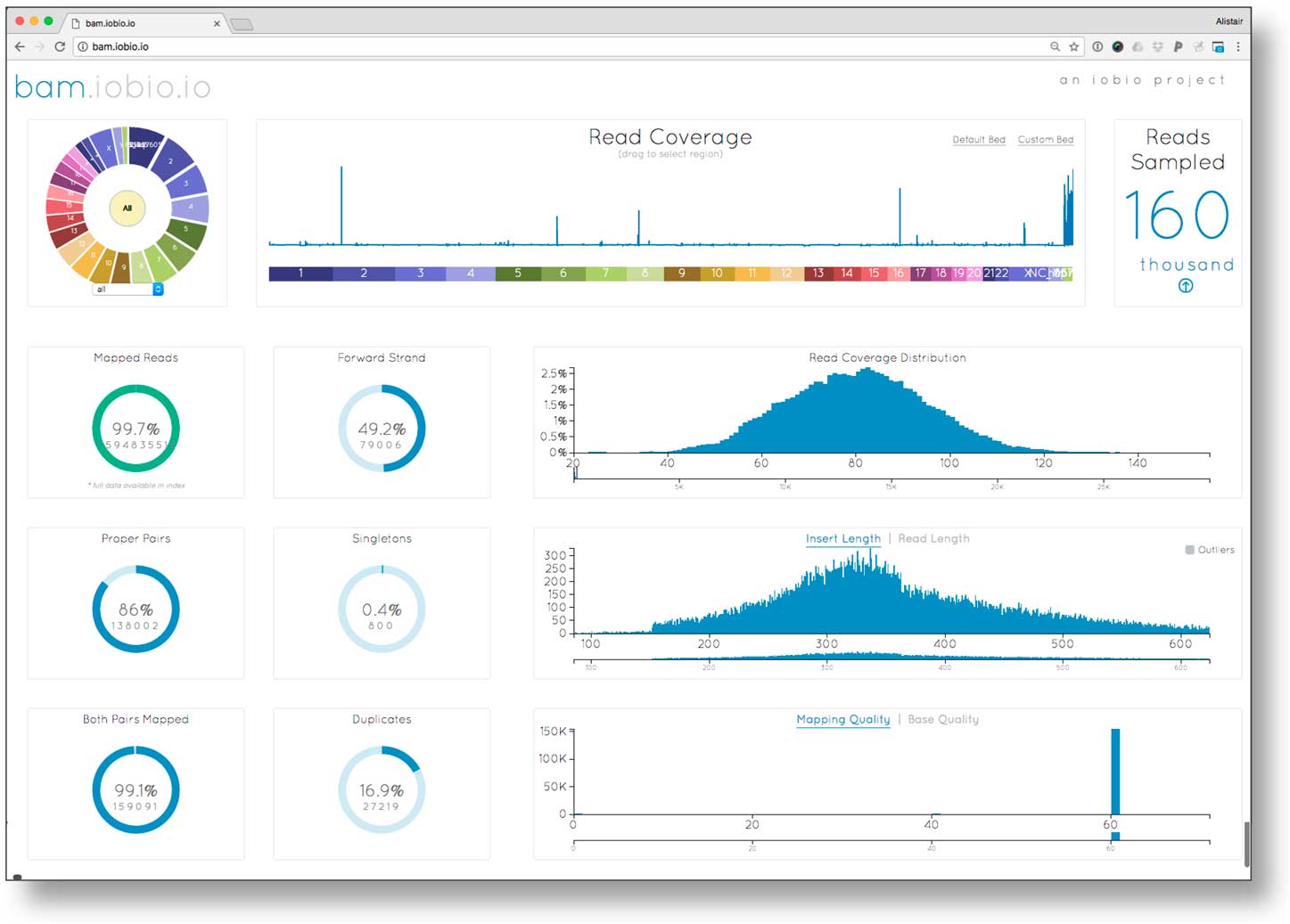
Fig. 1 Examining sequence alignment quality in the proband using the bam.iobio tool. Sequence coverage across all chromosomes (top middle), and relevant alignment metrics are visualized, including the distributions of read coverage, fragment length, and mapping quality (histograms on the right); as well as summary metrics including read mapping rate, and polymerase chain reaction (PCR) duplication rate (ring charts on left).
To search for the variant that caused the disease in the patient, we first defined the location of the sequence alignment (BAM) files from the 3 members of the family trio, then added a list of genes known to be associated with early childhood seizures using the Phenolyzer function. Next, due to the absence of defined variant call format [Reference Danecek16] files, the Freebayes variant calling algorithm, available natively within gene.iobio, automatically generated variant calls within this set of genes in about 2 minutes. Variants in the proband and 2 parents, together with the underlying sequence coverage, were then visualized (see Fig. 2).
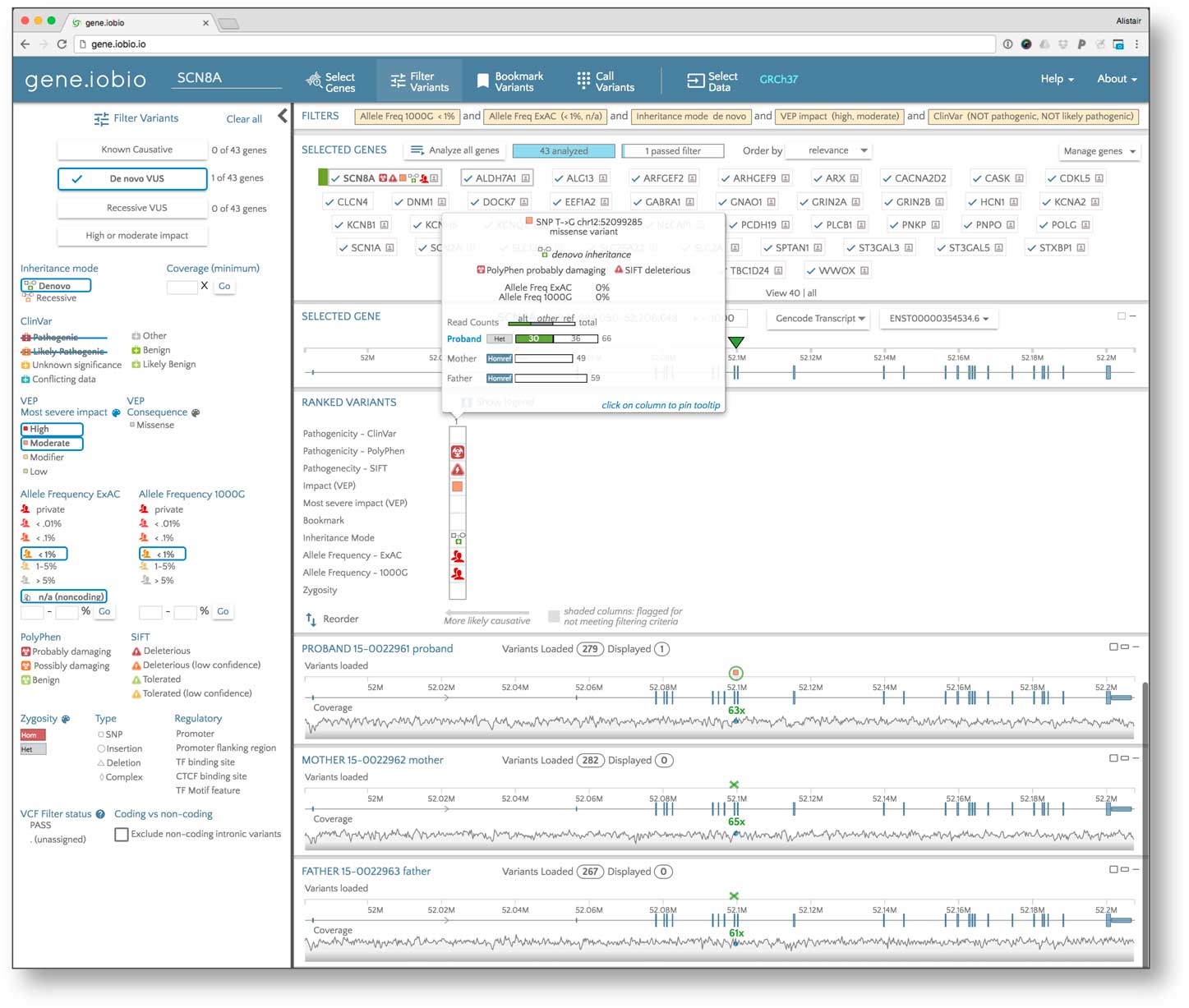
Fig. 2 Identifying the causative variant in the proband using the gene.iobio tool. This tool facilitates sample data selection (i.e., sequence alignment and variant files for the proband and parents); candidate gene list generation according to the patient phenotype; variant filtering according, for example, to mode of inheritance, observed and/or predicted pathogenicity, and population frequency; and gene/variant ranking and prioritization. The insert shows the salient properties of the diagnostic de novo disease-causing variant in the proband pinpointed by the tool.
In addition to generating the variant calls, gene.iobio also prioritized the variants using variant frequencies in the 1000 Genomes Project and the ExAC database [Reference Lek13] (updated to gnomAD [17] by the time of this publication; and noting that only variants with low population allele frequencies are likely to cause EIEE, a rare and usually dominant condition), predicted functional impact by the VEP prediction software [Reference McLaren18] (noting that variants with high predicted impact on function are more likely to be causative) and presence in ClinVar (noting that the causative variant may already be present in disease variant databases). This prioritization is presented in the “Ranked Variants” table (situated between the gene panel and sample data panel in the app), where the variants with the most significant effect appear to the far left. All of the genes defined in the initial panel appear at the top of the page with a visual summary of the high impact variants contained within them. Using this table, the highest ranked gene was determined to be SCN2A.
The highest ranked variant in SCN2A was a missense variant (ENST00000375437.2:c.647T>G) showing a de novo inheritance pattern with 0% allele frequency in the EXAC and 1000 Genomes databases. The variant is predicted by SIFT [Reference Ng and Henikoff19] to be deleterious, by PolyPhen [Reference Flanagan, Patch and Ellard20] as “probably damaging” and is marked in ClinVar as likely pathogenic. SCN2A encodes for the alpha subunit of a voltage-gated neuronal sodium channel known to be associated with EIEE [Reference Ogiwara21]. A quick analysis of the other highly ranked genes unearthed no other variants likely to be implicated in EIEE for this patient. The missense variant in SNC2A meets American College of Medical Genetics and Genomics criteria [Reference Richards22] as likely pathogenic and was deemed diagnostic for the subject. Subsequent confirmatory clinical testing with a second, EIEE-specific gene panel including the SCN2A gene validated this variant finding.
Discussion
We developed the iobio tool suite with the goal of enabling clinicians and researchers with minimal bioinformatics expertise and limited access to computational resources to rapidly perform complex genome analyses for diagnosis and discovery purposes. Iobio tools are web applications that spare the user from having to run UNIX command line programs, offer intuitive user interfaces, present results visually, and obviate the need for extensive training. These web tools are architected such that all analyses are carried out on “backend” servers currently hosted on Amazon Web Services (although local copies of the tools are also available upon request, to allow analysis of patient data securely behind institutional firewalls), and the web client is only responsible for the visual “rendering” of the results. Because the tools analyze only the small portion of the underlying genomic data that is immediately relevant, either sampling a representative fraction of the data to estimate critical statistics (e.g., read coverage), or targeting specific regions of a patient’s genome (e.g., a gene or subset of genes), both data analysis on the backend server and data transfer between the server and the client can be reduced to seconds, allowing the tools to return results in real time. Furthermore, because of the small amount of data that needs to be transferred, wired or wireless networks typically available in a hospital or research office are sufficient. Finally, because all analyses are performed server-side, iobio analyses can be carried out on laptop computers purchased within the past few years, that is without any “special” hardware. By reducing analysis time and eliminating the need for bioinformatics expertise and computational infrastructure, we aim to increase the relative cost-effectiveness of genome sequencing-based diagnoses and improve genetic diagnostic rates.
A number of effective variant investigation and prioritization tools are currently available in the commercial sector (Opal from Fabric Genomics [23], the Ingenuity Variant Analysis Tool [24], the Alamut software suite [25], WuXi NextCode’s clinical analysis software suite [26], Congenica’s Sapienta platform [Reference Estellon27]) and in the public domain (QueryOR [28], PhenIX [29], MedSavant [30]). Our iobio tools offer unique analysis capabilities not available from these tools. For example, iobio is the only variant prioritization tool that integrates access to the primary data (i.e., to the BAM format read alignments), allowing the analyst to examine the underlying sequencing data and uncover potential data quality issues. For example, in our gene.iobio tool, variants are displayed directly adjacent to the corresponding read coverage at that location, reducing the potential for making incorrect calls due to insufficient sequencing data. In addition, iobio tools uniquely offer variant calling on demand, enabling analysis before batched variant calling is complete. Furthermore, iobio is the only tool to offer real-time analysis capabilities beyond variant filtering and sorting, and a sophisticated visual interface promoting intuitive analysis with minimal training.
In this study, we used our iobio tool suite to analyze >60× WGS Illumina data from an EIEE patient that had previously received a nondiagnostic panel test for neurodevelopmental disorders involving seizures, and was then recruited into a research study. Our analyses uncovered the likely diagnostic variant in SCN2A. Though SCN2A is a well-characterized cause of EIEE, it was not included in the previous genetic testing for this patient because of her complex phenotypic presentation including epilepsy and hypotonia, and later by profound neurodevelopmental impairment. The case illustrates the difficulty in selecting the appropriate panel test for genetically and phenotypically heterogeneous conditions, even when epilepsy (EIEE) symptoms are a prominent feature. The mutation has since been confirmed, the family has received counseling, and treatment has proceeded accordingly.
We note that our iobio analyst was able to confirm data quality and identify the SCN2A mutation using a standard laptop within minutes of selecting the BAM files, bypassing the need for any large-scale computing. The analysis did not require the upload of large BAM files (because iobio only accesses the relevant portions of the data files from their storage location). Furthermore, the analyst was able to carry out the analysis without running a whole-genome variant calling pipeline (because iobio has the ability to call variants “on demand” in the regions of selected genes).
In the EIEE case presented here, 60× WGS data was available as part of the research study. Importantly, the large size of our data set compared with an exome or gene panel data set did not reduce the speed or ease of our iobio analysis. The physician’s expertise and familiarity with clinical information of the patient was especially helpful to narrow in on the most common genes and variants and to determine mode of inheritance. Although gene.iobio allows the analyst to highlight, for example X-linked dominant, recessive, or autosomal dominant variants, physician knowledge led us to search for de novo mutations in the patient presented above. Like a hypothesis-based panel test, gene.iobio operates by analyzing subsets of genes with a high likelihood of association with the disease, reducing the functional search space to cover the “usual suspects” first. However, unlike a panel test, if the causative variant is not discovered in the first pass, the search can be broadened immediately and iteratively to include any additional gene or subset of genes, rather than ordering another panel test. This is currently accomplished by the embedded Phenolyzer tool that can pull in additional genes potentially associated with a given condition, enabling discovery of novel genes and variants. Thus, the gene.iobio tool offers the advantages of a targeted, hypothesis-based panel approach, with the option to expand the search iteratively across the entire exome or genome when necessary.
Although our team included informatics experts who were able to help our clinicians, we believe that these tools are easy to utilize without help from such informatics specialists. In order to use iobio apps, a clinician needs access to their patient’s/research subject’s sequence alignment files (BAM [Reference Li6]) and/or VCF [Reference Danecek16] file(s). These files need to be indexed (a standard step performed in modern bioinformatics pipelines) to allow access to specified genomic regions (e.g., genes), and must either be accessible as a URL, or be stored locally on the clinician’s own computer. The “Files” tab in gene.iobio provides the interface to select the files for analysis. The GNomEx [Reference Nix8] genomic data LIMS at the our institution simplifies this process by providing clickable links for our physician/analysts to commence analysis on a given patient. Similar integration at the research/clinician’s own institution may provide substantial convenience. Although basic functionality in gene.iobio is intuitive, we have provided a host of educational materials to help with the many advanced features this tool offers: instructional videos are available on the gene.iobio landing page, and topical tutorials and explanatory blog posts are accessible from the tool’s “Help” tab.
The analysis presented here demonstrates that our novel iobio web tools are already effective for fast and intuitive diagnostic variant investigation and prioritization. We continue to develop these tools both to add new functionality and to improve ease of use for our clinician users. One planned improvement is the incorporation of functionality to evaluate variants of unknown significance according to the complex American College of Medical Genetics and Genomics criteria [Reference Richards22]. We suggest that clinicians using gene.iobio will be able to diagnose a large fraction of clinical cases quickly and conclusively, eliminating further rounds of testing for many patients and their families. Some fraction of analyses will reveal variants with clinically uncertain effects that must be functionally validated before impacting patient treatment. Though these findings may not be immediately actionable in the clinic, such novel discoveries will expand our understanding of disease etiology, guide translational research, and help future patients.
Acknowledgments
This work was supported by grants UL1TR001067-04S2 to W.D. and G.T.M. from the National Center for Advancing Translational Sciences; U01HG006513 from the National Human Genome Research Institute to G.T.M., and DP2MH100008 from the National Institute of Mental Health to J.L.B.
Disclosures
A.W., C.M., and G.T.M. are founders, owners, and officers of Frameshift Labs, Inc., a software company developing products using the IOBIO technology. Frameshift Labs, Inc., has an exclusive commercial licensing agreement with the University of Utah for the commercial use of the gene.iobio web application covered in this manuscript. All other authors have no conflicts of interest to declare.



 Open access
Open access