


Metabolic regulation in complex organisms relies partly on transcriptional control as a long-term mechanism affecting the level of expression of key enzymes(Reference Desvergne, Michalik and Wahli1). In rodents there is high correlation between mRNA expression of target genes and recruitment of lipogenic transcription factors or nuclear receptors and their co-regulatory proteins to promoter regions, suggesting that gene expression analysis is useful for inferring transcriptional activity(Reference Bennett, Seo and Datta2). Transcriptional regulation of hepatic lipogenic gene expression, adipogenesis, or skeletal muscle fatty acid oxidation in rodents is under the control of sterol regulatory element-binding factor 1 (SREBF1)(Reference Horton, Goldstein and Brown3), carbohydrate responsive element-binding protein (ChREBP; also referred to as MLX interacting protein-like (MLXIPL))(Reference Towle4), and the ligand-activated nuclear receptors PPARγ and PPARδ(Reference Fernyhough, Okine and Hausman5). Less is known regarding the molecular events during skeletal muscle growth in livestock species such as cattle and pigs(Reference Hausman, Dodson and Ajuwon6). Recent work in this area, however, has indicated that the level of dietary starch fed to young cattle can alter gene networks associated with adipocyte differentiation and energy metabolism(Reference Graugnard, Piantoni and Bionaz7). Whether early exposure to high-starch or high-fibre diets has long-term carryover effects on gene networks remains to be established.
High-starch–low-fibre diets, through shifting the pattern of endproducts of ruminal fermentation towards greater propionate and lactate, provide readily available sources of energy (i.e. glucose) for growing muscle, and often lead to greater feed conversion efficiency and rates of growth(Reference Myers, Faulkner and Nash8, Reference Schoonmaker, Fluharty and Loerch9). The central hypothesis of the present study was that metabolic gene networks in longissimus lumborum muscle tissue during rapid post-weaning growth would be altered to different extents by feeding diets during the growing phase that varied in level of starch but provided similar amounts of energy for gain. A gene network can be defined as a collection of DNA segments which interact either with a regulator such as a transcription factor or nuclear receptor, but also with each other through their RNA and protein products and with other molecules in the cell(Reference Graugnard, Piantoni and Bionaz7). These ‘global’ interactions in turn can govern the rates at which genes in the network are transcribed into mRNA(Reference Bennett, Seo and Datta2).
As an extension of our previous work on early-weaned cattle transcriptomics(Reference Graugnard, Piantoni and Bionaz7), specific objectives were to study mRNA expression during the growing phase of eleven of the fourteen genes associated with adipogenesis that we examined originally plus an additional four genes including for the first time the lipogenic transcription regulator MLXIPL (commonly referred to as carbohydrate responsive element-binding protein; ChREBP) and the adipogenic repressor nuclear receptor subfamily 2, group F, member 2 (NR2F2). In addition, we examined four genes encoding signal transduction mediators and two nuclear receptors associated with cellular energy metabolism. We have previously shown that longissimus lumborum biopsies, which represent a mixture of adipocytes and myocytes among other cell types, are appropriate to study adipogenesis. More importantly, they can be performed repeatedly on the same animal over different stages of growth, are minimally invasive, and circumvent the need to kill the animal (see Graugnard et al. (Reference Graugnard, Piantoni and Bionaz7)).
In addition to our focus on gene networks, an important objective also was to examine potential ‘metabolic imprinting’(Reference Srinivasan and Patel10) effects, i.e. long-term up-regulation or down-regulation of metabolic genes due to early exposure to high-starch diets, as well as carryover effects of dietary starch level. In order to achieve all our specific objectives, we biopsied longissimus lumborum before the start of the diets (day 0) and at 56, 112 and 224 d on the experimental diets. Thus, unlike previously(Reference Graugnard, Piantoni and Bionaz7), the present study extended the evaluation of adipogenic and energy metabolism gene networks through the finishing phase when all animals had been fed the same high-starch diet for about 112 d.
Materials and methods
Experimental animals, diets and sampling
The study utilised fourteen early-weaned (age range of 155 ± 10 d at weaning) purebred Angus steers from the University of Illinois beef cattle herd. The steers were randomly assigned to a high-starch (HiS; 5980 kJ/kg diet DM; n 7) or a low-starch (LoS; 4970 kJ/kg diet DM; n 7) diet for 112 d. Dietary treatments in the present study were specifically designed to provide contrasting levels of starch and fibre while providing sufficient energy, i.e. calculated net energy for gain in diets HiS and LoS differed by about 20 % but was adequate in both cases to support ≥ 1·5 kg body weight per d. Both diets were formulated to be isonitrogenous. The LoS–high-fibre diet contained (g/kg DM): 350 maize silage; 200 maize gluten feed; 380 soya hulls; 30 cracked maize; 30 soyabean meal (490 g/kg crude protein). The HiS–low-fibre diet contained (g/kg DM): 200 maize silage; 680 cracked maize; 110 soyabean meal (490 g/kg crude protein). Both diets contained (g/kg DM): 10 limestone/dicalcium phosphate/mineral/vitamin/urea/dry molasses mixture. Calculated dietary fibre content was 5·9 % with HiS and 24 % with LoS. At the end of the growing phase, steers on each group were gradually adapted to a common high-grain finishing diet containing 6030 kJ calculated net energy for gain per kg DM. All diets were offered on an ad libitum basis. Steers had an individual electronic identification ear tag and individual feed intake data were collected using the GrowSafe® system (GrowSafe Systems Ltd, Alberta, Canada). Steer weights were recorded on consecutive days before treatment (i.e. a range in age of 155 ± 10 d), and at 56, 112 and 224 d after the start of the treatments. Individual animal average daily gain and daily DM intake were used to estimate feed conversion efficiency (gain:feed, kg/kg; Table 2). All steers received an implant of 20 mg oestradiol benzoate and 200 mg progesterone (Component® E-S; Vetlife Inc., West Des Moines, IA, USA) after the biopsy on day 56. Ultrasound images of longissimus lumborum area were captured at 112, 166 and 224 d of the growing phase using a 500 V Aloka (Corometrics Medical Systems, Inc., Wallingford, CT, USA) ultrasound with a 3·5 MHz transducer fitted to a custom beef animal standoff. Data were analysed with AUSkey System Software (Animal Ultrasound Services, Ithaca, NY, USA). Commercial vegetable oil was applied to the site of measurement to decrease sound wave attenuation associated with hair coat.
Biopsies and blood samples
Muscle biopsies were collected at 0, 56, 112 and 224 d relative to the start of feeding treatment diets (i.e. a range in age of 155 ± 10 d). Tissue was obtained from longissimus lumborum via needle biopsy (12 gauge core biopsy needle Bard® Magnum®; C. R. Bard Inc., Covington, GA, USA) while animals were immobilised in a cattle chute. The surgical area was clipped with fine clippers and washed with an iodine disinfectant mixture. Lidocaine-HCl (3 ml; Agri Laboratories, St Joseph, MO, USA) was given intramuscularly to anaesthetise the biopsy area before performing a 1 cm incision with a sterile scalpel blade. The first biopsy was collected from a section between the 12th and 13th rib on the left side of the animal. Subsequent biopsies were collected from the left side about 6 cm from the previous one moving towards the head. Over 0·5 g of tissue was obtained from each steer at each time point and was stored in liquid N2 until RNA extraction. The incision was then closed with surgical staples (Multi-Shot Disposable Skin Stapler, 3M Medical Products; Henry Schein®, Melville, NY, USA) and iodine ointment (Povidone ointment, 10 %; Henry Schein®, Melville, NY, USA) was applied to the wound. Animals were monitored daily for behavioural signs of discomfort and wound swelling or discharge. Staples typically fell off as the wound healed and few remained by 7 d post-biopsy, when those remaining were removed.
Blood was collected from the jugular vein before biopsies (about 08.00 hours) to isolate serum for metabolites following standard protocols at the Veterinary Diagnostics Laboratory, College of Veterinary Medicine, University of Illinois. Serum insulin concentration was quantified using a commercial bovine insulin ELISA kit (catalogue no. 10-1201-01; Mercodia AB, Uppsala, Sweden). Animals had free access to feed and consumed about six meals per d, thus minimising the potential for postprandial effects on blood metabolite concentrations.
RNA extraction, PCR, and primer design and evaluation
Biopsy tissue was weighed (about 0·3–0·5 g) and immediately subjected to RNA extraction using ice-cold Trizol (Invitrogen Corp., Carlsbad, CA, USA) as described previously(Reference Loor, Dann and Everts11). Genomic DNA was removed from RNA with DNase using RNeasy Mini Kit columns (Qiagen, Hilden, Germany). RNA concentration was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The purity of RNA (A260/A280) for all samples was above 1·9. RNA quality was assessed using a 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). Samples had a median RNA integrity value (RIN) of 7·3 (sd 0·2). The RIN is an algorithm for assigning integrity values to RNA measurements. This algorithm is applied to electrophoretic RNA measurements and assigns values from 1 to 10 (low to high quality) based on the 18S and 28S rRNA area and the height of the 28S rRNA peak (Agilent Technologies, Inc., Santa Clara, CA, USA). A portion of the RNA was diluted to 100 mg/l using DNase/RNase-free water before RT.
cDNA was synthesised using 100 ng RNA, 1 μg dT18 (Operon Biotechnologies, Huntsville, AL, USA), 1 μl 10 mm-dNTP mix (Invitrogen Corp.), 1 μl random primers (Invitrogen Corp.) and 10 μl DNase/RNase-free water. The mixture was incubated at 65°C for 5 min and kept on ice for 3 min. A total of 6 μl of master mix composed of 4·5 μl 5X First-Strand Buffer, 1 μl 0·1 m-dithiothreitol, 0·25 μl (50 U) of SuperScript™ III RT (Invitrogen Corp.) and 0·25 μl of RNase Inhibitor (10 U; Promega, Madison, WI, USA) was added. The reaction was performed in an Eppendorf Mastercycler® Gradient using the following temperature program: 25°C for 5 min, 50°C for 60 min and 70°C for 15 min. cDNA was then diluted 1:4 (v/v) with DNase/RNase-free water.
Quantitative PCR (qPCR) was performed using 4 μl diluted cDNA combined with 6 μl of a mixture composed of 5 μl 1 × SYBR Green master mix (Applied Biosystems, Foster City, CA, USA), 0·4 μl each of 10 μm forward and reverse primers and 0·2 μl DNase/RNAse-free water in a MicroAmp™ Optical 384-Well Reaction Plate (Applied Biosystems). Each sample was run in triplicate and a six-point relative standard curve plus the non-template control were used (User Bulletin no. 2; Applied Biosystems). The reactions were performed in an ABI Prism 7900 HT SDS instrument (Applied Biosystems) using the following conditions: 2 min at 50°C, 10 min at 95°C, forty cycles of 15 s at 95°C (denaturation) and 1 min at 60°C (annealing and extension). The presence of a single PCR product was verified by the dissociation protocol using incremental temperatures to 95°C for 15 s plus 65°C for 15 s. Data were calculated with the 7900 HT Sequence Detection Systems Software (version 2.2.1; Applied Biosystems). The final data were normalised using the geometric mean of the four most stable genes among the ones tested as internal controls, as reported previously(Reference Bionaz and Loor12).
Design and evaluation of primers
Primer features for those genes not reported previously by Graugnard et al. (Reference Graugnard, Piantoni and Bionaz7) are shown in Table 1. Primers were designed using Primer Express 2·0 or 3·0 with minimum amplicon size of 80 bp (when possible amplicons of 100–150 bp were chosen) and limited 3′ G+C (Applied Biosystems). When possible, primers were designed to fall across exon–exon junctions. Primers were aligned against publicly available databases using BLASTN (Basic Local Alignment Sequence Tool For Nucleic Acid) at the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) and the University of California Santa Cruz's Cow (Bos taurus) Genome Browser Gateway(13). Before qPCR, primers were tested in a 20 μl PCR reaction using the same protocol described for qPCR except for the final dissociation protocol. For primer testing we used a universal reference cDNA (RNA mixture from five different bovine tissues) to ensure identification of the desired genes. A quantity of 5 μl of the PCR product was run in a 2 % agarose gel stained with ethidium bromide (2 μl). The remaining 15 μl were cleaned using the QIAquick® PCR Purification Kit (Qiagen) and sequenced at the Core DNA Sequencing Facility of the Roy J. Carver Biotechnology Center at the University of Illinois (Urbana-Champaign, IL, USA). Only those primers that did not present primer-dimer, a single band at the expected size in the gel, and had the right amplification product (verified by sequencing) were used for qPCR. The accuracy of a primer pair also was evaluated by the presence of a unique peak during the dissociation step at the end of qPCR.
Table 1 GenBank accession number, sequence and amplicon size of primers used to analyse gene expression by quantitative PCR*

MLXIPL, MLX interacting protein-like; NR2F2, nuclear receptor subfamily 2, group F, member 2; ADIPOQ, adiponectin; ACSM1, acyl-CoA synthetase medium-chain family member 1; PDK4, pyruvate dehydrogenase kinase, isoenzyme 4.
* Information for the remainder of the genes has already been published (see Graugnard et al. (Reference Graugnard, Piantoni and Bionaz7)).
† Primer direction and hybridisation position on the sequence.
‡ Exon–exon junctions are underlined.
Selection and evaluation of internal control genes
Special attention was given to the selection and evaluation of reference genes for normalisation of PCR data; complete details of these procedures have been published previously(Reference Graugnard, Piantoni and Bionaz7). The geometric mean of single-stranded interacting protein 2 (RBMS2), ribosomal protein S15a (RPS15A), ubiquitously-expressed transcript (UXT) and mitochondrial GTPase 1 homologue (MTG1) was used to normalise gene expression data.
Statistical analysis
Body weight, growth performance, blood metabolites and insulin, and PCR data were analysed using the MIXED procedure in SAS (SAS Institute Inc., Cary, NC, USA). Before statistical analysis, normalised PCR data were transformed to fold-change relative to day 0 (i.e. before animals were started on experimental diets)(Reference Graugnard, Piantoni and Bionaz7). To estimate standard errors at day 0 and prevent biases in statistical analysis, normalised PCR data were transformed to obtain a perfect mean of 1·0 at day 0, leaving the proportional difference between the biological replicate. The same proportional change was calculated at all other time points to obtain a fold-change relative to day 0. Fixed effects in the statistical model for each variable analysed (i.e. genes, blood metabolites, performance) included diet, day on experiment, and diet × day on experiment when appropriate (i.e. for metabolites and mRNA expression over time). Gene expression and blood metabolite and insulin data analysis included a repeated-measures statement with an autoregressive covariate structure. The random effect in all models was steer within diet. All means were compared using the PDIFF statement of SAS (SAS Institute, Inc., Cary, NC, USA). Significance was declared at P ≤ 0·05.
Clustering analysis
Hierarchical clustering was performed using fold-changes in mRNA expression for each diet on day 56, day 112 and day 224 relative to day 0 using Genesis software (Genesis, Bournemouth, Dorset, UK)(Reference Sturn, Quackenbush and Trajanoski14). The standard correlation coefficient, i.e. the dot product of two expression vectors, provides an indication of the degree of co-expression between genes(Reference Eisen, Spellman and Brown15). In hierarchical clustering, relationships among objects (genes) are represented by a tree whose branch lengths reflect the degree of similarity between the objects, as assessed by a pairwise similarity function(Reference Eisen, Spellman and Brown15) such as that described above. Euclidean distance was used as the measure of similarity in the behaviour of genes in the present study. The clustering algorithm used is based closely(Reference Sturn, Quackenbush and Trajanoski14) on the average-linkage method developed to compute a dendrogram that assembles all elements into a single tree(Reference Eisen, Spellman and Brown15).
Gene network analysis
Summary networks among genes were developed using the web-based software package Ingenuity Pathway Analysis® (www.ingenuity.com; Redwood City, CA, USA). The networks were generated using the respective gene identifiers and the actual fold-changes in expression in the comparison HiS v. LoS at 56, 112 and 224 d on treatments. Only those genes affected significantly (P ≤ 0·05) by the interaction of diet × time are highlighted. Connections among genes were based on known relationships available in the Ingenuity Pathway Analysis® knowledge base. This is a proprietary manually-curated database containing relationships from the published literature in rodents and humans.
All experimental procedures involving steers were approved by the University of Illinois Institutional Animal Care and Use Committee under protocol no. 05095.
Results and discussion
Overall animal performance
Steers in the present study consumed increasing amounts of feed during the growing phase (Fig. 1), a response probably associated with increased concentrations of plasma glucose and insulin (Fig. 2). In general, the HiS diet was most efficacious in terms of growth and performance during the growing phase (Table 2), confirming previous results(Reference Graugnard, Piantoni and Bionaz7, Reference Schoonmaker, Cecava and Faulkner16, Reference Schoonmaker, Cecava and Fluharty17). High-starch v. low-starch diets fed to early-weaned Angus × Simmental steers (nineteen or twenty animals per diet) for 100 d resulted in moderately greater intramuscular fat (3·6 v. 3·2 %) measured via ultrasound(Reference Schoonmaker, Cecava and Faulkner16). Our steers fed LoS, however, consumed greater overall amounts of energy than those fed LoS in the study of Schoonmaker et al. (Reference Schoonmaker, Cecava and Faulkner16), which probably explains the lack of statistical significance on marbling scores at the end of the growing phase (565 v. 518 for HiS and LoS). We observed similar responses in a recent study with both early-weaned Angus and Angus × Simmental steers(Reference Graugnard, Piantoni and Bionaz7).

Fig. 1 Daily DM intake (DMI) (a and b) and estimated net energy for gain (NEG) intake (c and d) (seven animals per diet) during the growing (a and c) and finishing (b and d) phases. Steers fed the low-starch diet (- -○- -) had greater (diet P = 0·03) DMI (about 9·4 v. 8·1 kg/d) during the growing phase than steers fed the high-starch diet (–●–). There was an overall effect of time (P < 0·001) and an interaction effect (P < 0·01) due to gradual increases in DMI over time with both diets. Estimated NEG intake, however, did not differ (P = 0·48) due to diet but increased over time regardless of diet (diet × time P < 0·001). During the finishing phase, neither DMI nor NEG differed (P = 0·91).

Fig. 2 Blood serum concentrations of NEFA (A), glucose (B) and insulin (C) in Angus steers (seven animals per diet) fed a high-starch (HiS; –●–) or low-starch (–○–) diet for a 112 d growing phase followed by a common HiS diet for an additional 112 d. Values are means, with standard errors represented by vertical bars. * Mean value was significantly different from that of the HiS diet (P < 0·05). a,b,c Mean values with unlike letters were significantly different (P < 0·05). An overall time effect (P < 0·05) was observed for NEFA, glucose and insulin.
Table 2 Performance and ultrasound measures of fat deposition in Angus steers (seven animals per diet) fed a high-starch (HiS) or low-starch (LoS) diet during a 112 d growing phase followed by a common HiS diet during a 112 d finishing phase
(Mean values and pooled standard errors)
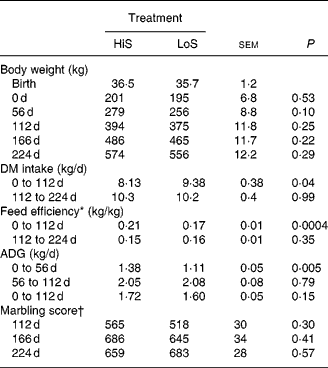
ADG, average daily gain.
* ADG/DM intake.
† Marbling score, where small = 500 to 599, modest = 600 to 699.
Gene markers of adipogenesis
In rodents, terminal differentiation of adipocytes requires up-regulation of mRNA of fatty acid-binding protein 4 (FABP4), glucose-6-phosphate dehydrogenase (G6PD), fatty acid synthase (FASN) and acetyl CoA carboxylase α (ACACA), among others, all of which are under control of PPARγ(Reference Hausman, Dodson and Ajuwon6, Reference Obregon18). Bovine perimuscular pre-adipocytes induced to differentiate with insulin and glucocorticoids in vitro had greater mRNA abundance of PPARγ, SREBF1, FABP4, acyl-CoA synthetase long-chain family member 1, and FASN after 2 d in culture compared with control(Reference Taniguchi, le Guan and Zhang19). Further, PPARγ and FABP4 expression remained elevated through 8 d in culture, suggesting that both were essential to sustain the differentiation programme or that they were abundantly expressed in mature adipocytes(Reference Taniguchi, le Guan and Zhang19).
Increased adipocyte number (i.e. proliferation) in the present study encompassing steers about 4–8 months of age is supported by some of the measured genes which are highly abundant in adipose compared with other tissues in mammals (for example, PPARγ, diacylglycerol O-acyltransferase homologue 2 (DGAT2), FABP4, stearoyl-CoA desaturase Δ9 desaturase (SCD))(Reference Hausman, Dodson and Ajuwon6). As in a previous study from our group(Reference Graugnard, Piantoni and Bionaz7), we observed marked increases in PPARγ (Fig. 3) and genes encoding lipogenic enzymes (ATP citrate lyase (ACLY), G6PD, FASN, glycerol-3-phosphate acyltransferase, mitochondrial (GPAM), FABP4, acyl-CoA synthetase medium-chain family member 1 (ACSM1), SCD and DGAT2) as well as the adipokine adiponectin (ADIPOQ) during the first 56 d of the growing phase exclusively due to greater dietary starch (Figs. 4 and 5). Previous studies reported a positive effect of ADIPOQ on lipogenesis in the pig and mouse(Reference Combs, Pajvani and Berg20, Reference Ajuwon and Spurlock21). Overall, the global expression patterns observed at 56 d in these animals closely resembled those at 112 d, i.e. they clustered very tightly (Fig. 6), providing an indication of sustained adipocyte differentiation.

Fig. 3 Patterns of mRNA expression of transcription regulators in longissimus lumborum tissue from Angus steers (seven animals per diet) fed a high-starch (HiS; –●–) or low-starch (–○–) diet for a 112 d growing phase followed by a common HiS diet for an additional 112 d: (A) PPARγ (PPARG); (B) sterol regulatory element-binding transcription factor 1 (SREBF1); (C) insulin-induced gene 1 (INSIG1); (D) MLX interacting protein-like (MLXIPL); (E) nuclear receptor subfamily 2, group F, member 2 (NR2F2); (F) thyroid hormone responsive (SPOT14 homologue, rat) (THRSP). Values are means, with standard errors represented by vertical bars. * Mean value was significantly different from that of the HiS diet (P < 0·05). a,b,c,d Mean values with unlike letters were significantly different (P < 0·05). There was an overall time effect (P < 0·05) for all genes. An overall diet effect (P < 0·05) was observed for THRSP and tendencies (P < 0·09) for PPARG and INSIG1.

Fig. 4 Patterns of mRNA expression of adipogenic and lipogenic enzymes in longissimus lumborum tissue from Angus steers (seven animals per diet) fed a high-starch (HiS; –●–) or low-starch (–○–) diet for a 112 d growing phase followed by a common HiS diet for an additional 112 d: (A) ATP citrate lyase (ACLY); (B) glucose-6-phosphate dehydrogenase (G6PD); (C) fatty acid synthase (FASN); (D) glycerol-3-phosphate acyltransferase, mitochondrial (GPAM); (E) fatty acid-binding protein 4 (FABP4); (F) acyl-CoA synthetase medium-chain family member 1 (ACSM1); (G) stearoyl-CoA desaturase (Δ9 desaturase) (SCD); (H) diacylglycerol O-acyltransferase homologue 2 (mouse) (DGAT2). Values are means, with standard errors represented by vertical bars. * Mean value was significantly different from that of the HiS diet (P < 0·05). a,b,c,d Mean values with unlike letters were significantly different (P < 0·05). There was an overall time effect (P < 0·05) for all genes. An overall diet effect (P < 0·05) was observed for ACLY, FABP4 and DGAT2.

Fig. 5 Patterns of mRNA expression of signal transduction mediators, nuclear receptors and co-activators and an adipokine in longissimus lumborum tissue from Angus steers (seven animals per diet) fed a high-starch (HiS; –●–) or low-starch (–○–) diet for a 112 d growing phase followed by a common HiS diet for an additional 112 d: (A) insulin receptor (INSR); (B) insulin receptor substrate 1 (IRS1); (C) PPARδ (PPARD); (D) pyruvate dehydrogenase kinase, isoenzyme 4 (PDK4); (E) adiponectin (ADIPOQ); (F) PPARγ, coactivator 1-α (PPARGC1A). Values are means, with standard errors represented by vertical bars. * Mean value was significantly different from that of the HiS diet (P < 0·05). a,b,c Mean values with unlike letters were significantly different (P < 0·05). An overall time effect (P < 0·05) was observed for all genes. An overall diet effect (P < 0·05) was observed for PDK4.

Fig. 6 Hierarchical clustering of gene expression patterns using Genesis software(Reference Sturn, Quackenbush and Trajanoski14) for each diet combination on days 56, 112 and 224 of the experiment relative to day 0. For all panels, the x axis corresponds to steers fed the high-starch (HiS) diet at day 56 (HiS56), the HiS diet at day 112 (HiS112), the low-starch (LoS) diet at day 56 (LoS56), the LoS diet at day 112 (LoS112), the HiS diet at day 224 (HiS224) and the LoS diet at day 224 (LoS224). White dots denote peak gene expression for each specific gene and diet combination. The relative degree of up-regulation (grey-to-black) relative to day 0 is based on the colour intensity. The length of the branches represents the relative degree of similarity for all genes within a diet and day combination (i.e. an entire column) or between genes across all diet combinations. MLXIPL, MLX interacting protein-like; ACLY, ATP citrate lyase; DGAT2, diacylglycerol O-acyltransferase homologue 2 (mouse); FASN, fatty acid synthase; ACSM1, acyl-CoA synthetase medium-chain family member 1; G6PD, glucose-6-phosphate dehydrogenase; ADIPOQ, adiponectin; GPAM, glycerol-3-phosphate acyltransferase, mitochondrial; INSIG1, insulin-induced gene 1; PPARG, PPARγ; PDK4, pyruvate dehydrogenase kinase, isoenzyme 4; NR2F2, nuclear receptor subfamily 2, group F, member 2; INSR, insulin receptor; IRS1, insulin receptor substrate 1; PPARD, PPARδ; PPARGC1A, PPARγ, coactivator 1-α; SREBF1, sterol regulatory element-binding transcription factor 1; FABP4, fatty acid-binding protein 4; SCD, stearoyl-CoA desaturase (Δ9 desaturase); THRSP, thyroid hormone responsive (SPOT14 homologue, rat).
Clustering analysis (Fig. 6) revealed that ACLY as well as DGAT2 and FASN (among the top four genes clustering together) might have been central to sustain the lipogenic process both by providing cytosolic citrate (ACLY) through lactate utilisation(Reference Whitehurst, Beitz and Pothoven22, Reference Whitehurst, Beitz and Cianzio23) and channelling of acetyl-CoA towards de novo fatty acid synthesis (FASN) and esterification (DGAT2). In general, clustering analysis revealed biologically relevant groupings of genes that sustained the pro-adipogenic effect of high dietary starch (for example, ACSM1, G6PD, GPAM). Further bioinformatics analysis with Ingenuity Pathway Analysis® (Fig. 7) underscored the fact that adipogenic genes responding to HiS by 56 d encompassed a closely regulated network (at least in non-ruminants) controlling adipocyte differentiation at an early age. However, contrary to our previous observations we found that steers fed the LoS diet experienced a marked increase between 56 and 112 d in the expression of lipogenic genes and particularly FASN, FABP4, SCD, and DGAT2 (Fig. 4). This seemingly ‘delayed’ lipogenic response explains the fact that global gene expression patterns on day 112 for these steers clustered in a group along with data at 56 and 112 d from steers fed HiS (Fig. 6). Taken together, the above changes were suggestive of high dietary starch causing pre-adipocytes to exit the proliferative phase and enter terminal differentiation at an earlier stage.

Fig. 7 Summary gene network expression in steers fed high-starch (HiS) v. low-starch (LoS) diets during a 112 d growing phase followed by a common HiS diet for an additional 112 d. (A) HiS v. LoS at 56 d; (B) HiS v. LoS at 112 d; (C) HiS v. LoS at 224 d. The relationships depicted are from the most current information in the Ingenuity Pathway Analysis® knowledge base. Shown at each time point are up-regulated (black background) and down-regulated (white background) genes with a significant diet × time interaction (according to Figs. 3–6) in the comparison of HiS- v. LoS-fed steers. Genes with a grey-coloured background were not affected significantly by the interaction of diet × time. ACLY, ATP citrate lyase; ACSM1, acyl-CoA synthetase medium-chain family member 1; ADIPOQ, adiponectin; DGAT2, diacylglycerol O-acyltransferase homologue 2 (mouse); FABP4, fatty acid-binding protein 4; FASN, fatty acid synthase; GPAM, glycerol-3-phosphate acyltransferase, mitochondrial; G6PD, glucose-6-phosphate dehydrogenase; INSIG1, insulin-induced gene 1; INSR, insulin receptor; IRS1, insulin receptor substrate 1; MLXIPL, MLX interacting protein-like; NR2F2, nuclear receptor subfamily 2, group F, member 2; PDK4, pyruvate dehydrogenase kinase, isoenzyme 4; PPARD, PPARδ; PPARG, PPARγ; PPARGC1A, PPARγ, coactivator 1-α; SCD, stearoyl-CoA desaturase (Δ9 desaturase); SREBF1, sterol regulatory element-binding transcription factor 1; THRSP, thyroid hormone responsive (SPOT14 homologue, rat); ![]() , enzyme;
, enzyme; ![]() , kinase;
, kinase; ![]() , ligand-dependent nuclear receptor;
, ligand-dependent nuclear receptor; ![]() , transcription regulator;
, transcription regulator; ![]() , transporter; ○, unknown; —, direct relationship; - - -, indirect relationship.
, transporter; ○, unknown; —, direct relationship; - - -, indirect relationship.
We are not aware of studies with cattle that have evaluated negative regulators of adipogenesis; thus, we examined potential regulators of PPARγ-driven adipogenesis (for example, Fig. 7). Work in rodent pre-adipocytes showed that upstream promoter-transcription factor II (COUP-TFII (COUP transcription factor 2), also known as NR2F2 (nuclear receptor subfamily 2, group F, member 2)) is a potent anti-adipogenic factor through the recruitment of the nuclear receptor 2 co-repressor complex (NCOR2) to binding sites close to both promoters of PPARγ(Reference Xu, Yu and Hsu24, Reference Okamura, Kudo and Wakabayashi25). This complex maintains the chromatin in a hypoacetylated and repressed state that is normally reversed upon differentiation(Reference Kinlaw, Church and Harmon26). In this context, it was puzzling that expression of key adipogenic enzymes studied (for example, FABP4, SCD, DGAT2) was greater in response to LoS at 112 and 224 d of feeding (Fig. 4), and that of PPARγ was greater at 224 d of feeding LoS (Fig. 3). Because overexpression of NR2F2 in vitro led to blunted up-regulation of FABP4, ADIPOQ, lipoprotein lipase (LPL), solute carrier family 2 (facilitated glucose transporter), member 4 (SLC2A4; insulin-stimulated glucose transporter) and PPARγ, all of which reduced rodent adipocyte differentiation(Reference Xu, Yu and Hsu24), it seems possible that responses due to LoS might not necessarily have led to greater adipogenesis. The physiological ligand of NR2F2 in non-ruminants is not known and it is difficult to speculate what might have triggered the response in animals fed LoS.
Although our focus was on gene networks, the expression pattern of thyroid hormone responsive (SPOT14 homologue, rat) (THRSP) (Fig. 3) deserves special attention due to its known role in lipogenesis in non-ruminants, its short-term regulation by thyroid hormone and insulin(Reference Obregon18, Reference Kinlaw, Church and Harmon26, Reference LaFave, Augustin and Mariash27), and also its long-term regulation via the transcription factor MLXIPL (Reference Towle4). The responses induced by LoS on THRSP expression were puzzling because in a previous study we found a marked increase of THRSP by 56 d of feeding Angus steers high-starch or low-starch diets(Reference Graugnard, Piantoni and Bionaz7). In that study, expression remained elevated (about 15- to 25-fold v. day 0) to 112 d regardless of starch level. From a physiological perspective it could be possible that this represented a compensatory mechanism to achieve a desired level of intramuscular fat deposition, i.e. other adipogenic or lipogenic genes remained at a lower expression level than steers fed HiS.
The strict requirement of SREBF1 for adipogenesis and lipogenesis in cattle is unknown. A recent study in rodents found that lipogenic gene expression in adipose tissue was independent of SREBF1(Reference Sekiya, Yahagi and Matsuzaka28). The marked up-regulation of insulin-induced gene 1 (INSIG1) expression by 56 d relative to SREBF1 (Fig. 3), if it extended to the protein level, might have resulted in reduced activity of SREBF1 because the primary function of INSIG1 in non-ruminant adipose tissue or liver is to block processing of SREBF1(Reference Li, Takaishi and Cook29, Reference Kast-Woelbern, Dana and Cesario30). Up-regulation of INSIG1 transcription in adipocytes down-regulated expression of PPARγ and SREBF1. The net result of INSIG1 action is the control of pre-adipocyte differentiation(Reference Li, Takaishi and Cook29), thus providing a feedback mechanism to restrict both lipogenesis and adipogenesis.
In rodents, MLXIPL is activated (i.e. phosphorylated) by glucose via the formation of xylulose-5-phosphate through the pentose phosphate pathway(Reference Towle4). In the present study, we observed that feeding HiS resulted in markedly greater MLXIPL (about 11-fold) by 56 and 112 d relative to day 0 (Fig. 3). Such pattern of expression closely mirrored serum glucose and insulin (Fig. 2), but also G6PD (Fig. 4), which encodes the rate-limiting enzyme of the pentose phosphate pathway. Expression of MLXIPL in HiS-fed steers was nearly 40-fold greater at 224 d relative to day 0 and about 10-fold greater compared with steers fed LoS. More importantly, up-regulation of MLXIPL would have allowed for sustained lipogenesis via its direct effects on target genes such as FASN and SCD (Fig. 7). It remains to be determined if this transcription factor also might be regulated by glucose or its metabolites in growing cattle.
Metabolic imprinting of adipogenesis due to dietary starch
The molecular mechanisms underlying postnatal programming effects due to altered nutrition in non-ruminants are largely unknown. It has been speculated that sustained hyperinsulinaemia in the postnatal period can result in lasting malprogramming of neuroendocrine systems that are critical for regulation of metabolism(Reference Srinivasan and Patel10). The increase in expression of SREBF1 and MLXIPL between 112 and 224 d in steers fed HiS was suggestive of a potential ‘metabolic imprinting’(Reference Srinivasan and Patel10) effect elicited by early exposure to high dietary starch, leading to further enrichment(Reference Rosen and MacDougald31) of intramuscular adipocytes at a more mature age of the animal. A recent study reported that hypermethylation of PPARγ in rodent preadipocytes prevents transcription of the gene and induction of differentiation(Reference Fujiki, Kano and Shiota32). Furthermore, adipocytes from diabetic mice have greater methylation of the PPARγ promoter coupled with lower mRNA abundance, all of which provided evidence that adipogenic transcription regulators (and potentially enzymes) might be regulated via epigenetic mechanisms(Reference Fujiki, Kano and Shiota32). Further detailed studies will be required to determine potential epigenetic regulation of adipogenic and lipogenic genes in cattle fed high-starch diets at an early age.
Gene markers of energy metabolism in myocytes
The present observations indicated that between 112 and 224 d, the decrease in serum insulin in steers fed HiS during the growing phase probably allowed for greater release of esterified long-chain fatty acids (Fig. 2) and/or reduced uptake of NEFA by muscle such that expression of both PPARδ and PPARγ, coactivator 1-α (PPARGC1A) remained unchanged, thus potentially allowing cells to sustain fatty acid oxidation for ATP synthesis. An important component of the glucose–fatty acid oxidation cycle is pyruvate dehydrogenase kinase, isoenzyme 4 (PDK4), which encodes a kinase that specifically phosphorylates the pyruvate dehydrogenase complex leading to inhibition of pyruvate oxidation to acetyl-CoA(Reference Kwon and Harris33). Gene expression and activity of PDK4 is under acute control by insulin (via FOXO1, also known as forkhead box O1(Reference Kwon and Harris33)) and under long-term control by PPARδ and PPARGC1A(Reference Kleiner, Nguyen-Tran and Bare34). Furthermore, PDK4 is a known PPARγ target gene in non-ruminants(Reference Berger and Moller35).
In tissues such as liver and skeletal muscle, PDK4 is particularly important during the starved state, not only to allow continued fatty acid oxidation, but also to conserve lactate, alanine and pyruvate for hepatic gluconeogenesis(Reference Kwon and Harris33). The marked increase in PDK4 expression between 56 and 112 d of feeding HiS (Fig. 5) might have led to accumulation of pyruvate in myocytes primarily. In that case, greater PDK4 expression coupled with greater serum insulin (Fig. 2) and insulin receptor (INSR) expression would have allowed for sustained fatty acid oxidation and sparing of glucose for glycogen synthesis(Reference Kwon and Harris33). In contrast, an accumulation of pyruvate in adipocytes of steers fed HiS as a result of PDK4 up-regulation could have stimulated fatty acid synthesis from both lactate and acetate produced during ruminal digestion(Reference Whitehurst, Beitz and Pothoven22, Reference Whitehurst, Beitz and Cianzio23).
Summary and conclusions
Exposure to HiS diets during the early growing phase appeared to induce precocious pre-adipocyte differentiation and lipid filling through up-regulation of PPARγ and its target genes (Fig. 7). HiS also led to greater SREBF1 and MLXIPL during the finishing phase, which coupled with greater G6PD and ADIPOQ could help sustain and promote adipogenesis. The LoS–high-fibre diet apparently delayed adipocyte differentiation as indicated by increased THRSP, FASN, FABP4, SCD and DGAT2 during the second half of the growing phase. An additional factor implicated in this delay was an apparent decrease in insulin sensitivity, indicated by the higher blood NEFA and similar insulin as well as unchanged insulin receptor (INSR). Quite surprisingly, animals fed the LoS–high-fibre diet during the growing phase had additional increases in expression of THRSP, PPARγ, FABP4, ACSM1, SCD and DGAT2 during the finishing phase. However, the concomitant increase in NR2F2 expression (a repressor of adipocyte differentiation in rodents) might have blunted the expected adipogenic response. The sustained up-regulation of MLXIPL and SREBF1 due to feeding high dietary starch at an early age was suggestive of metabolic imprinting.
Acknowledgements
We gratefully acknowledge the help from T. G. Nash and J. Dahlquist as well as the rest of the staff at the University of Illinois Beef Cattle Unit for animal handling and care.
D. E. G. collected skeletal muscle biopsies and blood, performed RNA extraction, selected internal control genes for qPCR, and performed qPCR analysis and data transformation. L. L. B., D. B. F. and J. J. L. conceived, designed and participated in the coordination of all aspects of the study. D. E. G. and J. J. L. drafted the manuscript. All authors read and approved the final manuscript.
There are no financial or other contractual agreements that might cause conflict of interests.









