


There has been increasing interest in the past few years on the bioactive compounds present in seaweeds( Reference MacArtain, Gill and Brooks 1 – Reference Brown, Allsopp and Magee 3 ). Traditionally, seaweeds are consumed as a food product in Asian countries and are increasingly used worldwide as ingredients for industrial applications. In Japan, over twenty species of red, green and brown algae (seaweed) are included in meals( Reference Arasaki and Arasaki 4 ), and daily seaweed consumption per person has remained relatively consistent over the past 40 years, in the range of 1·50–3·65 kg/person per year, as reported by a range of studies( Reference Zava and Zava 5 – Reference Matsumura 7 ). Seaweeds are a rich source of polyphenolic compounds( Reference Koivikko, Eranen and Loponen 8 ), and polyphenols extracted from algae( Reference Dutot, Fagon and Hemon 9 , Reference Wijesinghe and Jeon 10 ) show some similarities to those found in land plants( Reference Dutot, Fagon and Hemon 9 – Reference Ragan and Craigie 11 ). Thus, the main polyphenols found in brown seaweeds are phlorotannins( Reference Glombitza, Hauperich and Keusgen 12 – Reference Glombitza and Zieprath 15 ), a type of phenolic compound only found in brown seaweeds( Reference Pal Singh and Bharate 16 ). Brown seaweed phlorotannins are oligomers and polymers of phloroglucinol units, and their oligomer and polymer molecular weights can greatly vary, from 126 Da to 650 kDa( Reference Brown, Allsopp and Magee 3 ), comprising up to 15 % of the plant dried weight( Reference Ragan and Craigie 11 ). It has been reported that the consumption of brown algae is on average 1·342 kg/person per year, containing 66·652 g of phlorotannins/person per year and 183 mg/person per d( Reference Arasaki and Arasaki 4 ). Phlorotannins are classified according to the type of linkages between phloroglucinol units into four main groups: eckols (with dibenzodioxin linkages), fucols (with a phenyl linkage), fuhalols and phloroethols (with ether linkages), and fucophloroethols (with ether and phenyl linkages)( Reference Pal Singh and Bharate 16 ). Phlorotannins are being increasingly investigated for their vast array of bioactivities( Reference Wijesinghe and Jeon 10 , Reference Kim and Himaya 17 , Reference Tsujimoto, Yokota and Vilcek 18 ) such as antioxidant( Reference Kang, Cha and Wijesinghe 19 – Reference Queguineur, Goya and Ramos 24 ), anti-inflammatory( Reference Kim, Shin and Lee 20 , Reference Jung, Jin and Ahn 25 , Reference Kim, Ku and Lee 26 ), antibacterial( Reference Eom, Kim and Lee 27 , Reference Biswas, McClure and Jimenez 28 ), anticancer( Reference Nwosu, Morris and Lund 29 – Reference Wheeler, Catravas and Odoms 33 ) and antidiabetic( Reference Nwosu, Morris and Lund 29 , Reference Steevensz, Mackinnon and Hankinson 34 , Reference Jegou, Kervarec and Cerantola 35 ), showing promising potential to further develop seaweed-derived products rich in bioactive components with commercial potential for food and pharma applications( Reference Wijesinghe and Jeon 36 ).
Bioavailability is a critical factor influencing in vivo biological activity of polyphenols and we have reasonable understanding of the bioavailability of polyphenols from fruits and vegetables, and some of the mechanisms by which they exert beneficial effects in vivo have been determined( Reference Corona, Vauzour and Amini 37 ). Their ability to act as effective bioactive molecules in vivo is dependent on the extent of their biotransformation( Reference Queguineur, Goya and Ramos 24 ) and conjugation during absorption from the gastrointestinal (GI) tract, in the liver and finally in cells( Reference Corona, Vauzour and Amini 37 ). Consequently, consideration must be given to the way polyphenols are absorbed and metabolised during GI digestion and colonic fermentation, and how this may have an impact on bioactivity( Reference Vauzour, Rodriguez-Mateos and Corona 38 ). It is noteworthy that there is no information on the bioavailability of seaweed phlorotannins, and this is a limitation to understanding their bioactivity and mechanism of action in vivo. In the absence of specific data regarding phlorotannin absorption and bioavailability, it is useful to consider the absorption and metabolism of other polyphenols as a guide( Reference Corona, Vauzour and Amini 37 ). In general, after ingestion of a polyphenol-rich diet, their protective effects in vivo are determined by measuring a range of suitable biomarkers, and they correlate with the absorption of polyphenols from the gut and their circulation/excretion( Reference Vauzour, Rodriguez-Mateos and Corona 38 ). Polyphenols can be extensively conjugated to form glucuronide, sulphate and methyl group in the gut mucosa and inner tissues( Reference Corona, Vauzour and Amini 37 , Reference Manach, Scalbert and Morand 39 ), and absorption occurs in the small intestine( Reference Corona, Vauzour and Amini 37 ). Polyphenols unabsorbed in the upper GI tract or re-excreted in the bile are extensively metabolised by colonic microflora into a wide range of low-molecular-weight phenolic acids( Reference Scalbert, Morand and Manach 40 ). The aim of this study was to elucidate the GI modifications of seaweed phlorotannins, and the effects on their metabolism and bioavailability. A food-grade seaweed polyphenol extract (SPE) rich in phlorotannins (from the brown seaweed Ascophyllum nodosum) was subjected to in vitro GI digestion and fermentation to examine the GI modifications occurring in the upper and lower GI tract. Furthermore, the absorption and metabolism of polyphenols in healthy subjects was investigated, after oral ingestion of a SPE capsule containing 101·89 mg of polyphenols. This amount represents an intake lower than the average daily intake of seaweed polyphenols in the Asian diet, and it is not expected to exert any cytotoxic effect( Reference Simmons-Boyce, Purcell and Nelson 41 ). The impact of absorption and GI modifications on phlorotannins anti-inflammatory potential is explored.
Methods
Seaweed material
Fresh A. nodosum was supplied by The Hebridean Seaweed Company, Isle of Lewis, Scotland in March 2011. The seaweed biomass was harvested by hand, cleaned and then shipped refrigerated to the processing facility in France where it was immediately chopped and frozen.
Preparation of food-grade seaweed polyphenol extract and capsule
A novel SPE from A. nodosum was produced by CEVA (France) using a solvent-based extraction system that was specifically developed for this study and for use with either fresh or frozen A. nodosum. The solvent used was a 60:40 ethanol–water mixture, which allowed for the water content of the seaweed itself. The extraction was carried out over 5 h using constant stirring and at all times protected from light. A solvent–seaweed ratio of 3:1 was used. The mixture was filtered to remove the supernatant, and subsequently the alcohol was removed using a rotary evaporator. A hydrometer was used to check that all of the alcohol had been removed. The final extract was recovered by centrifugation and further filtration before freeze-drying.
Approximately half of the produced extract (basic extract) was then fractionated using tangential flow ultrafiltration to produce further extracts of varying molecular-weight range and with varying polyphenol content. A blended SPE was formulated (Table 1) using 175 mg of basic extract and 50 mg of high-molecular-weight (HMW) fraction (>10 kDa cut-off) for use in the current study. Maltodextrin (175 mg) was added to the capsule formulation as an excipient. This was done in order to maximise the polyphenol content (>100 mg/d) but also to minimise the level of I to within accepted regulatory guidelines (<500 μg/d). Blending was carried out at the food-grade CEVA facilities in France. Doses of 400 mg of the SPE Ascophyllum blend were packed into white, opaque, vegetarian capsules by Irish Seaweeds, Belfast, UK and used for the clinical study. The food-grade seaweed capsule was characterised by normal-phase (NP)-HPLC (NP-HPLC) and liquid chromatography (LC)-MS analysis. Phlorotannins were quantified using the Folin–Ciocalteu Method( Reference Singleton 42 ) using phloroglucinol as the standard( Reference Koivikko, Eranen and Loponen 8 ).
Table 1 Key components of polyphenol-rich basic extract, high-molecular-weight (HMW) fraction and blend (capsule) showing crucial concentrations of polyphenols and iodine
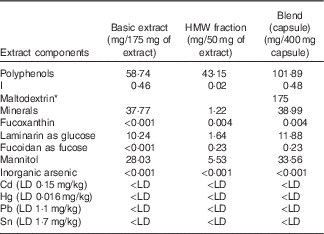
LD, limit of detection.
* Maltodextrin was added to the capsule formulation as an excipient.
Simulated gastrointestinal digestion and fermentation
The GI digestion procedure was adapted from Mills et al. ( Reference Mills, Tuohy and Booth 43 ) (2008) and McDougall et al.( Reference McDougall, Dobson and Smith 44 ) (2005). This method consists of two sequential stages: gastric digestion and small intestinal digestion followed by dialysis. A measure of 10 g of SPE was dissolved in 30 ml of acidified water (pH=2), and pepsin (320 U/ml) was added. Samples were incubated at 37°C for 2 h on a shaker covered with foil to protect them from light. Aliquots of 5 ml (G) were removed. The pH was adjusted to 7·5 by adding a few drops of 6 m-NaOH, and pancreatin (4 mg/ml) and bile extracts (25 mg/ml) were added. The samples were incubated at 37°C for 2 h on a shaker. Aliquots of 5 ml (SI) were removed. Samples were transferred into the dialysis tubing (100–500 Da, cut-off, 1·8 ml/cm, Spectra/Por; Biotech) and dialysed overnight at 4°C against water (4 litres) to remove low-molecular-weight digests. Aliquots of 5 ml of dialysis solution (D1) were removed. The dialysis fluid was changed and dialysis was continued for an additional 2 h. Aliquots of 5 ml of second dialysis solution (D2) were removed. Samples (SI+D) were freeze-dried and subjected to colonic fermentation (batch culture): the method was adapted from Tzounis et al.( Reference Tzounis, Vulevic and Kuhnle 45 ) Batch-culture fermentation vessels (300 ml; one vessel per treatment) were autoclaved and filled with 135 ml of sterilised basal medium. Medium was stirred and gassed overnight with O2-free N2. Before the addition of SI+D-digested extracts equivalent to 1·5 g of undigested extracts, the temperature inside the vessels was set to 37°C by a circulating water bath, and the pH was controlled at 6·8 by an Electrolab pH controller (Electrolab UK), in order to mimic conditions in the distal region of the human large intestine (anaerobic; 37°C; pH 6·8). Vessels were inoculated with 15 ml of faecal slurry (1:10, w/v), and batch cultures were run for 24 h. Samples measuring 7 ml were collected at five time points (0, 2, 4, 8 and 24 h), centrifuged at 13 000 rpm at 4°C for 10 min and the supernatants were kept. All the samples collected during the digestion and fermentation procedures were stored at −80°C until LC-MS analysis.
Study design
This study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects/patients were approved by the University of Reading Ethics Committee before initiation of the study. Written informed consent was obtained from all participants. Exclusion criteria for subjects were as follows: smokers, BMI<18 or >30 kg/m2, abnormal liver function and haematology, alcohol intake of >21 U/week, GI disease or chronic GI disorders, consumption of antibiotics in previous 3 months before study and women who were pregnant or intending to become pregnant. Potentially suitable participants underwent a screening process, and individuals with blood pressure >150/90 mmHg; Hb>125 g/l for men and >110 g/l for women, γ-glutamyl transferase >1·3 mkat/l or cholesterol>6·5 mmol/l were excluded from the study. In total, twenty-four volunteers were recruited: twelve female volunteers (six aged 18–30 years and six aged 30–65 years) and twelve male volunteers (six aged 18–30 years and six aged 30–65 years). Participants were asked to follow a low phenolic diet for 1 d before the study day (devoid of tea, coffee, fruit, vegetables, alcoholic beverages, cocoa, whole-grain and seaweed-containing products). On the day of the study, the subjects were cannulated and a baseline blood sample was taken. Participants were asked to consume one SPE capsule (400 mg) containing 101·89 mg of polyphenols. Blood samples were collected at 0, 1, 2, 3, 4, 6, 8 and 24 h after ingestion of the SPE capsule, and urine samples were collected at baseline, 0–8 and 8–24 h after the ingestion. During the day, participants were provided with a lunch and dinner of low phenolic content. The study is registered at clinicaltrials.gov (study ID: NCT02496806).
Sample collection and storage
One aliquot of blood was collected in heparin tubes and cultured immediately (whole blood culture for cytokine analysis). One aliquot of blood was collected in EDTA tubes and centrifuged at 3000 rpm for 15 min at 4°C. The plasma was separated and 1 mg/ml ascorbic acid was added as preservative. Aliquots were stored at −80°C until analysis. Total volume of collected urine was recorded, and aliquots were stored at −80°C until analysis.
Plasma sample processing for metabolite analysis
Plasma samples were prepared by following a procedure similar to the one described by Ottaviani et al.( Reference Ottaviani, Momma and Heiss 46 ). A volume of 10 µl of internal standard solution (resorcinol 200 µg/ml) was added to 450 µl of plasma, and then 50 µl of 1·2 m-acetic acid was added and samples were mixed. Samples were analysed with and without enzymatic treatment (37°C, 40 min) in the presence of 1500 IU of β-glucuronidase and 50 IU of sulfatases from Helix pomatia (type H-1). A volume of 1 ml of 100 % methanol acidified with 0·5 % acetic acid was added, and samples were centrifuged for 15 min at 16 100 g at 4°C and supernatants were collected. This step was repeated three times (last time with 50 % methanol acidified with 0·5 % acetic acid), and the supernatants were dried using a SpeedVac. The pellets were dissolved with 125 µl of mobile phase and transferred to vials for reverse-phase (RP)-HPLC (RP-HPLC) analysis.
Urine sample processing for metabolite analysis
Urine samples were prepared according to a procedure similar to the one described by Ottaviani et al.( Reference Ottaviani, Momma and Heiss 46 ). Briefly, 10 µl of internal standard solution (resorcinol 200 µg/ml) was added to 250 µl of urine. Samples were analysed with and without enzymatic treatment (37°C, 40 min), in the presence of 1500 IU of β-glucuronidase and 50 IU of sulfatases from H. pomatia (type H-1). A volume of 1 ml of 100 % methanol acidified with 0·5 % acetic acid was added, samples were mixed and centrifuged for 15 min at 16 100 g at 4°C, and supernatants were transferred to a new tube and dried on a SpeedVac. Dried samples were re-suspended on 125 µl of mobile phase, completely dissolved, centrifuged and transferred to vials for RP-HPLC analysis and LC-MS analysis.
Normal-phase HPLC analysis
The phlorotannins in the food-grade SPE used to produce the capsule were analysed by NP-HPLC analysis( Reference Koivikko, Eranen and Loponen 8 ) using an HPLC 1100 series equipped with LiChrospher Si60-5 column (250 mm×4·0 mm ID, 5 μm particle size from Hichrom (LISP60-5-250AF)), fitted with a guard column LiChrospher Si60-5 from HICHROM (LISP60-5-10C5). The mobile phase contained A: 82 % dichloromethane+2 % methanol+2 % acetic acid in water and B: 96 % methanol+2 % acetic acid in water, and it was pumped through the column at a rate of 1 ml/min. A volume of 10 µl of samples was injected and analysed by the gradient programme, which was (min/%B) as follows: 0/0, 30/17·6, 45/30·7, 50/87·8, 60/87·8, 80/0, 105/0 for detection of all compounds. The compounds were detected at a wavelength of 268 nm. All data were analysed by the ChemStation software. The phloroglucinol standard was injected at 0·1–100 µg/ml, and phlorotannins in the capsules were analysed as phloroglucinol equivalents.
Reverse-phase HPLC analysis
The analysis of plasma and urine samples was carried out with a Hewlett-Packard 1100 series liquid chromatograph (Hewlett-Packard), as previously described( Reference Corona, Tzounis and Assunta Dessi 47 ). Samples were analysed by RP-HPLC using a Nova-Pak C18 column (4·6×250 mm) with 4 µm particle size. The temperature of the column was maintained at 30°C. The mobile phases consisted of a mixture of 5 % aqueous methanol in 0·1 % hydrochloric acid 5 m (A) and a mixture of aqueous acetonitrile 50 in 0·1 % hydrochloric acid 5 m (B), and they were pumped through the column at a rate of 0·7 ml/min. The following gradient system was used (min/%B): 0/5, 5/5, 40/50, 55/100, 59·9/100, 60/5, with 10 min post-run for both compound and metabolite detections. The eluent was monitored by photodiode array detection at 280 nm and spectra of products obtained over the 200–600 nm range. Peaks were characterised by their retention time and spectra characteristics. A calibration curve of phloroglucinol was constructed using authentic standards (0·1–100 µg/ml), and in each case they were found to be linear with correlation coefficients of 0·995. Metabolites were quantified as phloroglucinol equivalents.
Liquid chromatography-MS analysis
LC-MS analysis was conducted to analyse the food-grade seaweed capsule, the urine samples and the digested materials, and it was carried out in the negative ion mode using LC-MS/MS utilising electrospray ionisation (ESI), as previously described( Reference Pinto, Paiva-Martins and Corona 48 ). Characterisation was achieved using LC-MS/MS utilising ESI. This consisted of an Agilent 1200 HPLC system equipped with a binary pump, degasser, auto-sampler, thermostat, column heater, photodiode array detector and an Agilent 1100 series LC/MSD Mass Trap Spectrometer (Agilent Technologies UK). Separation of samples was achieved using a Zorbax SB C18 column (2·1×100 mm; 1·8 µm) (Agilent), and HPLC conditions were as follows: injection volume, 1 µl; column temperature, 25°C; binary mobile system, (A) 0·1 % aqueous formic acid and (B) 0·1 % of formic acid in acetonitrile; and flow rate, 0·2 ml/min. A series of linear gradients were used for separation (min/%B): 0/10, 3/10, 15/40, 40/70, 50/70, 65/10. MS was performed in the negative ion mode (scan range, m/z 100–1000 Da; source temperature, 350°C). All solvents used were of LC-MS grade.
Cytokine production
Blood samples collected during the clinical intervention (baseline, 1, 2, 4, 6 and 8 h) into heparin tubes were immediately cultured as follows: 500-μl blood aliquots were mixed with 500 μl of Roswell Park Memorial Institute (RPMI) medium containing antibiotics on a twenty-four-well plate, and lipopolysaccharide (LPS) (1 μg/ml) or vehicle (control group) was added before incubation at 37°C for 24 h. At the end of the culture period, samples were centrifuged at 2000 g for 5 min, and the supernatants were collected and kept at −20°C until analysis for inflammatory cytokine levels. The supernatants were collected and stored at −20°C. Cytokines (IL-1β, IL-6, IL-8, IL-10 and TNF-α) in the supernatants were measured with Luminex xMAP Technology using commercially available Fluorokine MAP kits (R&D systems), and data were analysed on the xPONENT software. Final data are presented as the difference between LPS-treated and unstimulated control.
Statistical analysis
The statistical evaluation of the results was performed by one-way ANOVA, followed by a Bonferroni post hoc t test using GraphPad InStat version 5 (GraphPad Software). Significant changes are indicated as P<0·05.
Results
Seaweed polyphenol extract characterisation
The chromatogram (Fig. 1) illustrates the trace obtained by NP-HPLC with diode array detection after injecting a water solution of the SPE. The chromatogram shows a number of peaks (20–70 min) representing different HMW phlorotannins, the characteristic phenolics in brown seaweeds. Longer phlorotannin polymers, which consisted of more hydroxy groups, resulted in tighter attachment to the column material. Consequently, shorter compounds were released earlier than longer compounds. Owing to a lack of phlorotannin standards and the complexity of the oligomeric and polymeric forms, the calibration curve of phloroglucinol was used to quantify all the phlorotannins contained in the SPE as phloroglucinol equivalents. The SPE comprised a wide range of molecular weights of phloroglucinol derivatives with a total phlorotannin concentration of 312 µg/mg quantified as phloroglucinol equivalents. Further characterisation of the SPE was achieved with LC-MS/MS (Fig. 2) using ESI. The data were collected in a non-targeted manner, by acquiring full spectrum data in negative ion mode from m/z 100 to 1000. The data were then analysed by searching for the theoretical mono-isotopic masses corresponding to possible phlorotannin oligomers and the presence of ions (1–6), which could correspond to phlorotannins (Fig. 2). The ion 1 with [M-H]- at m/z 405 corresponded to the trimer hydroxytrifuhalol A, whereas the MS2 fragment −387 corresponded to the loss of one molecule of water (−18), a characteristic pattern of phlorotannin fragmentation. Compound 2 ([M-H]- at m/z 497) was considered to be a phlorotannin tetramer composed of 4 phloroglucinol units, such as tetrafucol or fucodiphlorethol, and also in this case the fragment −479 corresponds to the loss of a molecule of water (−18), whereas the fragment −353 corresponds to the loss of water (−18) and a phloroglucinol unit (−126), in accordance with analytical profiles recently described in positive ion mode by Wang et al.( Reference Wang, Jonsdottir and Liu 49 ) and by Ferreres et al.( Reference Ferreres, Lopes and Gil-Izquierdo 50 ). The ion 3 has a [M-H]- at m/z 247 corresponding to a C-O-C dimer of phloroglucinol, as previously indicated by Nwosu et al. ( Reference Nwosu, Morris and Lund 29 ). The ion 4 (387) corresponds to the trimer 7-hydroxyeckol, and we observe the presence of a fragment at −369 deriving from the loss of one molecule of water. Isomers 5 and 6 with [M-H]- at m/z 249 were also observed, which can correspond to the dimers diphlorethol and difucol.

Fig. 1 Chromatographic separation of phlorotannins contained in the seaweed extract by normal-phase HPLC with diode array detection (268 nm).

Fig. 2 Characterisation of phlorotannins in the seaweed extract. (a) Structures of phlorotannins identified in the seaweed extract. (b) Phlorotannins in the seaweed extracts identified by liquid chromatography (LC)-MS analysis in negative ion mode.
In vitro digestion and characterisation
Because of the lack of commercially available standards for phlorotannins and the complexity of the oligomers and polymers in the extract, the analysis of phlorotannins and their metabolites is challenging and requires a combination of approaches. Similarly to other polyphenol classes, phlorotannins may undergo extensive modification by phase I and phase II enzymes and the colonic microbiota during their transit through the GI tract( Reference Corona, Vauzour and Amini 37 ), and the implication of such metabolic modifications on the bioactivity of phlorotannins has not been investigated yet. Consequently, we subjected the SPE to in vitro digestive and fermentative processes. An in vitro gastric and ileal digestion and colon microbial fermentation of the SPE was initially conducted, followed by dialysis to simulate absorption into the circulation. The MS spectra and fragmentations of the compounds detected in the samples were studied (Fig. 3). After in vitro digestion and fermentation procedures, the samples were analysed by LC-MS/MS using ESI in negative ion mode, as previously described, searching for the theoretical mono-isotopic masses corresponding to the low-molecular-weight phlorotannins previously identified in the capsule (Fig. 2). We were able to identify molecular ions and fragments corresponding to hydroxytrifuhalol A (405), the C-O-C dimer of phloroglucinol (247), the dimer diphlorethol/ difucol (249) and 7-hydroxyeckol (387), also found after colonic fermentation. In addition, in digested and fermented samples subjected to dialysis to mimic absorption into the circulation, we reported the presence of seven compounds (digestion metabolite (DM)1–DM7) corresponding to in vitro-absorbed metabolites.

Fig. 3 Liquid chromatography (LC)-MS analysis in negative ion mode of the seaweed extract subjected to in vitro gastrointestinal digestion, colonic fermentation and dialysis to mimic absorption. (a) LC-MS spectra and fragmentation of in vitro-digested materials. (b) Summary of LC-MS analysis of the in vitro-digested materials. DM, digestion metabolite. SIM, small intestinal metabolite; FM, fermentation metabolite.
Plasma and urine analysis
The food-grade SPE was given to healthy subjects (Fig. 4) in the form of a capsule (400 mg) containing 101·89 mg of polyphenols, and blood and urine samples were analysed for phlorotannin metabolites. HPLC-diode array detector (DAD) analysis of the plasma (Fig. 5) and urine (Fig. 6) samples with and without glucuronidase/sulfatase treatment showed the presence of a variety of metabolites absent in the baselines (before the seaweed capsule ingestion) in samples from fifteen volunteers out of twenty-four. In plasma, the total level of phlorotannins and their metabolites ranges from 0·011 to 7·757 µg/ml, and in urine 0·15 to 33·52 µg/ml are excreted. Some metabolite peaks were present in samples with and without enzymatic treatment, and therefore could be assigned to unconjugated metabolites. Some other metabolite peaks were present only in samples without enzymatic treatment or were only appearing in samples enzymatically treated, and were attributed to conjugated forms (glucuronides and/or sulphates) and their enzymatically released unconjugated forms. In urine, some metabolites were found in samples collected at 0–8 h after capsule ingestion, but the majority of the metabolites were found in samples collected at 8–24 h. Some metabolites, such as urine metabolite (UM)6 and UM7, show similar UV spectra characteristics and might therefore be structurally related (Fig. 6). In plasma (Fig. 5), some metabolites were found in samples collected at 2, 3 and 4 h after capsule ingestion, but the majority of the metabolites were found in samples collected at later time points (6–24 h). This could be the result of poor absorption of the HMW phlorotannins in the upper GI tract, resulting in them reaching the colon and undergoing fermentation to lower-molecular-weight derivatives by the colonic microbiota. In addition, urine samples were subjected to LC-MS/MS (Fig. 7) using ESI, as previously described, searching for the theoretical mono-isotopic masses corresponding to the low-molecular-weight phlorotannins previously identified in the capsule (Fig. 2). We were able to identify molecular ions and fragments corresponding to hydroxytrifuhalol A, 7-hydroxyeckol and the C-O-C dimer of phloroglucinol, which corresponded to the HPLC metabolite UM3. In addition, we reported the presence of two ions (289 and 377) corresponding to metabolites that we characterised in samples from SPE subjected to in vitro GI digestion and fermentation (DM4 and DM7, Fig. 3), as previously detailed.

Fig. 4 Schematic illustration of the clinical intervention setup. SPE, seaweed polyphenol extract

Fig. 5 HPLC analysis of plasma samples for seaweed metabolites. (a) HPLC chromatograms (268 nm) and UV spectra showing examples of metabolites in plasma. (b) Summary of seaweed metabolites present in plasma samples. PM, plasma metabolite; RP-HPLC, reverse-phase HPLC.

Fig. 6 HPLC analysis of urine samples for seaweed metabolites. (a) HPLC chromatograms (268 nm) and UV spectra showing examples of metabolites in urine. (b) Summary of seaweed metabolites present in urine samples. UM, urine metabolite; RP-HPLC, reverse-phase HPLC.

Fig. 7 Liquid chromatography (LC)-MS analysis in negative ion mode of urine samples. (a) LC-MS spectra and fragmentation of phlorotannins found in urine samples. (b) Summary of LC-MS analysis of the urine samples. DM, digestion metabolite.
Ex vivo cytokine production
The ex vivo production of cytokines (IL-1β, IL-6, IL-8, IL-10 and TNF-α) relative to baseline levels in cultured blood collected a various time points (0, 1, 2, 3, 4, 6 and 8 h) during the intervention study (LPS treated – unstimulated controls) is reported in Fig 8. The amounts of TNF-α and IL-10 remained quite stable over time, as well as the amount of all cytokines at 1 and 2 h. IL-6 levels were observed to decrease at later time points (4–8 h) without reaching statistical significance (P>0·05). The levels of both IL-1β and IL-8 were observed to increase from 3 to 8 h after the intervention; however, the statistical analysis revealed that the only significant change from baseline levels was the increase of IL-8 at 8 h.

Fig. 8 Cytokine production by whole blood cultures in cultured blood collected at various time points (0, 1, 2, 3, 4, 6 and 8 h) during the intervention study (lipopolysaccharide treated – unstimulated controls). a
P<0·05 v. baseline. ![]() , IL-1β;
, IL-1β; ![]() , IL-6;
, IL-6; ![]() , IL-8;
, IL-8; ![]() , IL-10,
, IL-10, ![]() , TNF-α.
, TNF-α.
Discussion
Polyphenols are ubiquitously found in plants and comprise a major part of a daily human diet. Over the past 20 years, significant data have emerged with regard to the potential beneficial effects of several classes of phenolic compounds against a number of chronic diseases. In addition, a reasonable understanding has been gained of the bioavailability of many polyphenol classes, and this will be important for understanding the mechanisms by which they exert such benefits in vivo. The interest in marine sources of phenolic compounds is recent, and knowledge on phlorotannin bioavailability is still lacking. The analysis of phlorotannins is challenging because of the high range of molecular weight present, and their characterisation is complicated further by the lack of commercially available standards.
Chromatography techniques coupled to diode array and MS detection have been applied to the analysis of phlorotannins, and the advantages/disadvantages of their use are described by Steevensz et al. ( Reference Steevensz, Mackinnon and Hankinson 34 ). RP-HPLC is a separation mode that is commonly used for polyphenol separation; however, the very high polarity of phlorotannins would cause them to elute with little or no retention because of the lack of interaction with the nonpolar stationary phase( Reference Steevensz, Mackinnon and Hankinson 34 , Reference Pinto, Paiva-Martins and Corona 48 ). NP-HPLC is more advantageous for retaining compounds with very high polarity, and the NP-HPLC methodology developed by Koivikko was more suitable than RP-HPLC for the separation of phlorotannins from the brown algae Fucus vesiculosus ( Reference Steevensz, Mackinnon and Hankinson 34 , Reference Pinto, Paiva-Martins and Corona 48 ). Thus, we initially analysed the phlorotannins in our SPE by NP-HPLC using a method adapted from Koivikko et al. ( Reference Koivikko, Eranen and Loponen 8 ). As expected, the SPE comprised a wide range of molecular weights (20–70 min), with abundance of very HMW phlorotannins eluting at later retention time (50–70 min) in our normal-phase method. MS detection can provide higher sensitivity and be advantageous to identify specific phlorotannins in the extract without commercially available standards; however, NP-HPLC solvents such as dichloromethane are not suitable for MS analysis( Reference Kazakevich and LoBrutto 52 ), because they would result in poor ionisation and therefore significantly reduce sensitivity( Reference Koivikko, Loponen and Pihlaja 51 , Reference Yanagida, Murao and Ohnishi-Kameyama 53 ).
Nwosu et al.( Reference Nwosu, Morris and Lund 29 ) characterised a phlorotannin extract from A. nodosum by RP-HPLC using a C18 column: the bound sample eluted in a unresolved set of peaks, and with MS detection in negative ion mode they were able to assign the detected m/z spectra to a series of phlorotannin structures( Reference Nwosu, Morris and Lund 29 ). Ferreres et al.( Reference Ferreres, Lopes and Gil-Izquierdo 50 ) identified twenty-two different phlorotannins belonging to the eckol and fucophloroethol groups in four seaweed species belonging to the order Fucales (genus Cystoseira and Fucus), with RP-HPLC separation combined with DAD-ESI-multiple-stage MS detection( Reference Ferreres, Lopes and Gil-Izquierdo 50 ). By using an equivalent RP-HPLC separation method coupled to ESI-MS analysis in negative ion mode, we were able to identify some phlorotannin oligomers such as hydroxytrifuhalol A, tetrafucol, fucodiphlorethol, C-O-C dimmer of phloroglucinol, 7-hydroxyeckol, diphloretol and difucol. The fragmentation patterns of the oligomers that we identified are in agreement with some recently published data in the field( Reference Wang, Jonsdottir and Liu 49 , Reference Ferreres, Lopes and Gil-Izquierdo 50 ). Recently, Steevensz et al.( Reference Steevensz, Mackinnon and Hankinson 34 ) characterised the phlorotannins of five brown algae species by ultrahigh-pressure liquid chromatography operating in ‘mixed-mode’ (hydrophilic interaction liquid chromatography mode) combined with high-resolution MS. The methodology proposed by this research group proved to be accurate for profiling phlorotannins based on their degree of polymerisation, and it was demonstrated to be an effective separation mode for low-molecular-weight phlorotannins, up to 6 kDa( Reference Steevensz, Mackinnon and Hankinson 34 ).
Phlorotannin characterisation is a challenging and complex task, which is complicated by the lack of commercially available standard compounds; thus, chromatography separation coupled to MS detection can help to elucidate phlorotannin complexity, and its application to the analysis of clinical samples from feeding trials, as well as the use of simplified in vitro digestion systems, can help elucidate their beneficial health properties and the bioactive circulating forms. The SPE was subjected to in vitro–simulated GI digestion and fermentation, followed by dialysis to simulate as close as possible their absorption and biotransformation. The obtained materials were analysed by LC-MS for a comparative characterisation of the phlorotannin metabolites. LC-MA analysis of the digested and fermented SPE has indicated the presence of some oligomeric phlorotannins that are also present in the undigested SPE (hydroxytrifuhalol A, diphloretol/difucol, 7-hydroxyeckol, C-O-C dimer of phloroglucinol), in addition to a range of newly formed metabolites (DM1 to DM7). In vitro conditions are indeed a great tool, allowing a simpler and more convenient analysis, and our in vitro system predicted the formation of metabolites subsequently identified in urine.
Intervention studies have investigated the fate of polyphenols from land plants in the human body by measuring plasma concentrations and/or urinary excretion following intake from a food source. Many studies performed to investigate polyphenol bioavailability are based on the measurement of their excretion in urine and plasma by extraction, concentration and chromatographic separation/analysis, and focused on the detection of polyphenols and their metabolites in samples subjected or not to enzymatic treatment to release conjugate moieties such as glucuronic acid and sulphate groups( Reference Ottaviani, Momma and Heiss 46 , Reference Miro-Casas, Covas and Farre 54 ). For example, after ingestion of a polyphenol-rich meal, levels of phenolic compounds and conjugated metabolites can increase rapidly, achieving a peak concentration at approximately 1–2 h in plasma and urine, indicating small intestinal absorption, or peak at later time points (4–8 h in plasma, 8–24 h in urine), indicating large intestinal metabolism and subsequent absorption( Reference Rodriguez-Mateos, Rendeiro and Bergillos-Meca 55 ). In our study, the majority of phlorotannin metabolites were found in samples collected at late time points (6–24 h), indicating limited small intestinal absorption followed by gut microbial metabolism of the HMW phlorotannins in the large intestine.
In the upper GI tract, dietary polyphenols act as substrates for a number of enzymes, and they are subjected to extensive metabolism by glucosidase enzymes, phase I enzymes (hydrolysing and oxidising), such as cytochrome P450, and phase II enzymes (conjugating and detoxifying) found both in the small intestine and the liver( Reference Manach, Williamson and Morand 56 ). In particular, there is strong evidence for the extensive phase II metabolism (by UDP-glucuronosyltransferases, sulphotransferases) to yield glucuronides and sulphate derivatives. Indeed, there is evidence of efficient glucuronidation and sulphation of all classes of polyphenols to differing extents( Reference Manach, Williamson and Morand 56 ). Indeed, our results indicate that phlorotannin intake results in the formation of phase II conjugate metabolites (glucuronides, sulphates).
Further transformations can occur in the colon, in which the enzymes of the gut microbiota act to breakdown complex polyphenolic structures to smaller units, which may also be absorbed and further metabolised in the liver. Bacterial enzymes may catalyse many reactions including hydrolysis, dehydroxylation, demethylation, ring cleavage and decarboxylation, as well as rapid deconjugation( Reference Selma, Espin and Tomas-Barberan 57 ).
As predicted by the HMW range of phlorotannins in our SPE, high colonic metabolism seems to have occurred, following fermentation of HMW phlorotannins in the large intestine. By LC-MS analysis, we were able, for the first time, to confirm the presence of some phlorotannin oligomers in urine, more specifically hydroxytrifuhalol A, 7-hydroxyeckol and the C-O-C dimer of phloroglucinol. Interestingly, two of the urine metabolites (m/z 289 and 377) were present in the in vitro-digested samples (DM4 and DM7).
There were substantial differences between volunteers in the pattern of phlorotannin metabolites. Such inter-individual differences have been observed for other polyphenols and have been attributed to differences in gut microbiota composition and the expression of metabolising enzymes( Reference Brown, Allsopp and Magee 3 , Reference Rodriguez-Mateos, Rendeiro and Bergillos-Meca 55 ).
A secondary aim of our work has been to determine whether the SPE could modulate inflammatory events in the blood, following the absorption of phlorotannin metabolites and because of their presence in the circulation.
Polyphenols can exert numerous antioxidant and non-antioxidant functions of relevance in chronic disease development, and many of them have an important inflammatory component( Reference Yanagida, Murao and Ohnishi-Kameyama 53 ). In the present study, we observed an altered ex vivo production of IL-8, a low-molecular-weight cytokine produced by mononuclear phagocytes and other cell types, with significant increased levels of the cytokine after 8 h compared with baseline.
IL-8 is an important inflammatory factor of the cysteine-intervening amino acid-cysteine (CXC) chemokine family, involved in the amplification of inflammatory signals( Reference Harada, Sekido and Akahoshi 59 ). IL-8 secretion is induced by TNF-α through a transcriptional mechanism primarily regulated by NF-κB)( Reference Fujishima, Hoffman and Vu 60 ). Redox signalling mechanisms are known to play a role in the regulation of such events, with reactive oxygen species being able to promote IL-8 production and secretion( Reference Tsujimoto, Yokota and Vilcek 18 , Reference McCord 23 ), whereas oxygen radical scavengers are proven to inhibit IL-8 production in LPS-stimulated human whole blood( Reference DeForge, Fantone and Kenney 22 ). Dietary polyphenols such as catechins( Reference Wheeler, Catravas and Odoms 33 ) and curcumin( Reference Biswas, McClure and Jimenez 28 ) have also been shown to specifically interfere with IL-8 gene expression through inhibition of NF-κB activation( Reference Rahman, Biswas and Kirkham 61 ). In consequence, we would have expected circulating seaweed polyphenol metabolites to potentially be able to inhibit IL-8 secretion. Our results have given a preliminary indication that the cytokine IL-8 is a possible target for phlorotannin metabolites. However, a significant increase in IL-8 levels at 8 h after the intervention was observed, in parallel with the presence of phlorotannin metabolites in plasma and urine samples. A recent study from our group investigated the influence of a polyphenol-rich intervention on inflammation as primary outcome. A randomised, double-blind, placebo-controlled, cross-over acute intervention was conducted, and cytokine levels (IL-8) were measured with the same ex vivo experimental protocol. The results showed a time-dependent increase in IL-8 release compared with baseline, in accordance with our findings. Thus, the postprandial ex vivo IL-8 production was significantly attenuated by the intervention compared with the control, with a parallel appearance in the circulation of polyphenol metabolites. Our trial was a single-group interventional study primarily designed to investigate the bioavailability of seaweed phlorotannins: however, on the basis of this preliminary indication on their anti-inflammatory potential, not sufficient to draw any conclusion, a chronic placebo-controlled intervention has been conducted to investigate the anti-inflammatory effect in deeper detail.
The main limitations of this study arise from the phlorotannin complexity and lack of commercially available analytical standards, potentially leading to possible quantification inaccuracy as phloroglucinol equivalents. The lack of analytical standards also implies a limited capability for method development, especially for the analysis of plasma, urine and digested materials. In future, the availability of standards could allow a higher degree of method optimisation and the development of specific solid-phase ion procedures for sample clean-up and concentration.
The development of more recently explored analytical applications to phlorotannins, such as hydrophilic interaction liquid chromatography( Reference Melanson and MacKinnon 32 ) and NMR( Reference Jegou, Kervarec and Cerantola 35 ), could facilitate the development of more suitable protocols that could lead to full identification of metabolites and improvements in phlorotannin metabolite quantification. In addition, the bioacessibility of polyphenols in the form of a capsule/extract might differ greatly from the bioacessibility of the same compounds in a food matrix( Reference Bohn 62 ). Future work will be needed to determine the potential effects on bioavailability of different food matrices and also any effects of cooking and/or processing.
Nevertheless, the present work has for the first time started to shed light on the role of colonic biotransformation on phlorotannin bioavailability, and its implication for their health benefits in vivo. Our results provide a basis for further investigating the seaweed-derived bioactive components in the body after ingestion; this information is necessary to understand their mechanism of action in vivo.
Acknowledgements
This work is part of the SWAFAX project (no. 262519), funded by the European Commission under the Capacities Programme (FP7).
We would like to thank all the SWAFAX project partners for their contribution, including Marigot, Hebridean Seaweed, Mesosystem S.L., Cybercolloids and the University of Ulster.
G. C., J. P. E. S., P. Y. and I. R. designed the research. G. C. organised and coordinated all parts of the clinical trial and analytical work. S. H. overviewed the preparation of seaweed materials, extracts and capsules. P. A. aided running the clinical trial. Y. J. aided with the analysis of metabolites in plasma and urines. G. C. analysed and summarised all the data. G. C. drafted the manuscript, C. G., I. R., S. H. and P. Y. revised the manuscript. I. R. had primary responsibility for final content.
The authors declare that there are no conflicts of interest.









