


In the Mediterranean diet, which has been associated with a reduced risk of CVD(Reference Barzi, Woodward, Marfisi, Tavazzi, Valagussa and Marchioli1–Reference Trichopoulou, Orfanos and Norat4), olive oil is the main source of fat. Although a variety of factors have been related with the heart-healthy effect of this diet, there is broad agreement in attributing a major role in this beneficial effect to the polyphenols in virgin olive oil (VOO)(Reference Covas5). This effect may originate from the action of polyphenols on different biochemical pathways, or from synergism between the effects of polyphenols and fatty acids, particularly oleic acid (a MUFA)(Reference Perez-Jimenez, Delgado, Pérez-Martínez, López-Segura, Fuentes, Cortés, Lozano and López-Miranda6).
The polyphenol compound present in highest concentrations in VOO is hydroxytyrosol (3,4-dihydroxyphenylethanol; HT); its concentration varies depending on the variety of olive, geographical area, ripeness of the fruit and method of olive oil pressing, among other factors(Reference Romero, Medina, Vargas, Brenes and De Castro7). For example, VOO from Manzanilla variety contains 2·5–3 times as much HT (479·6 μmol/kg oil) as the Hojiblanca (159·2 μmol/kg oil), Picual (176·8 μmol/kg oil), Cornicabra (164·1 μmol/kg oil) or Arbequina (179·6 μmol/kg oil) varieties(Reference Romero, Medina, Vargas, Brenes and De Castro7).
The effect of polyphenols on physiopathological mechanisms involved in CVD has been characterized only in part. These effects include inhibition of platelet aggregation(Reference Petroni, Blasevich, Salami, Papini, Montedoro and Galli8, Reference Dell'Agli, Maschi, Galli, Fagnani, Dal Cero, Caruso and Boisio9), inhibition of oxidation of LDL(Reference Fitó, Covas, Lamuela-Raventós, Vila, Torrents, de la Torre and Marrugat10), stimulation of NO production(Reference Pignatelli, Di Santo, Buchetti, Sanguigni, Brunelli and Violi11) and down-regulation of the expression of endothelial adhesion molecules(Reference Dell'Agli, Fagnani and Mitro12). Platelet aggregation is considered one of the main events in arterial thrombosis, and this makes investigating how aggregation is inhibited by polyphenols in VOO a worthwhile topic for further inquiry.
Although HT is the major polyphenol on VOO, other polyphenols are present, including hydroxytyrosol acetate (HT-AC)(Reference Romero, Medina, Vargas, Brenes and De Castro7). The concentration of HT-AC is approximately equal to that of HT in some olive varieties such as Arbequina, but is twice as high as that of HT in the Picual variety, and three to four times as high in the Manzanilla and Hojiblanca varieties(Reference Romero, Medina, Vargas, Brenes and De Castro7).
The aims of the present study were to measure the antiplatelet aggregating effect in vitro of HT-AC in human blood, and to compare its effects to HT and acetylsalicylic acid (ASA), the most widely used drug in the world to prevent ischaemic CVD because of its antiplatelet aggregating action.
Experimental methods
Materials
Thromboxane B2, 6-keto-prostaglandin F1α (6-keto-PF1α) and TNFα enzyme immunoassay kits were from Amersham International plc (Little Chalfont, UK). Nitrite/nitrate and the 3-nitrotyrosine ELISA kits and HT were obtained from Cayman Chemical (Ann Arbor, MI, USA). HT-AC was supplied by the Department of Organic Chemistry and Pharmaceutics, School of Pharmacy, University of Seville (Seville, Spain). Collagen and ADP were obtained from Menarini Diagnóstica S.A. (Barcelona, Spain). All other reagents were from Sigma Chemical Co. (St. Louis, MO, USA).
Study design
The present in vitro study was carried out in blood samples from healthy volunteers who had not taken any medication during the 15 d before inclusion and VOO in the diet 24 h before. Sodium citrate (3·8 %) at a proportion of 1:10 was used as the anticoagulant. In each group of experiments we used six samples from different volunteers (three men and three women in each group) with a mean age of 32·8 (se 1·3) years. The study was approved by the Clinical Assays Committee of the Hospital Regional Universitario Carlos Haya in Malaga, Spain.
Samples were used as whole blood, but in some experiments, platelet-rich plasma (PRP), erythrocytes and leucocytes were isolated from whole blood. PRP was obtained after centrifugation of whole blood at 800 g for 10 min at 20°C; all PRP samples were adjusted to 3 × 109 platelets/l. Erythrocytes were obtained from the pellet resulting from centrifugation of whole blood to obtain PRP; erythrocytes were washed twice with PBS (pH 7·4). Leucocytes were separated on a Ficoll density gradient (Hystopaque 1077 and Hystopaque 1119) and diluted (final count: 5·2 (se 0·2) × 109 leucocytes) in Ca2+ physiological saline solution (95 % viability as judged by trypan blue exclusion).
Different concentrations of ASA, HT and HT-AC (10− 7 to 10− 3m) were incubated at 37°C before inducers were added. All drugs were diluted in a mixture of ethanol–PBS (100 μl ethanol plus 900 μl PBS) at a proportion of 1:10.
Analytical techniques
All techniques were run in a single-blind manner, i.e. the persons who did the assays were unaware of the origin and nature of the samples.
Platelet aggregometry
Platelet aggregation was measured as electronic impedance in whole blood, PRP, PRP plus erythrocytes, and PRP plus leucocytes (500 μl sample diluted with 500 μl PBS). We used a Chrono-Log 540 aggregometer (Chrono-Log Corp., Haverton, PA, USA) with collagen (1 μg/ml), ADP (2·5 μm) and arachidonic acid (400 μm) to induce aggregation. Drugs were incubated at 37°C for 10 min before the aggregation inducer was added, and aggregation was recorded for 10 min. Maximum intensity of aggregation was quantified as the maximum change in electronic impedance in samples without the drug or a given concentration of each drug. The concentrations of aggregating agents were chosen according to previous experiments which yielded EC50 values of 1·10 (se 0·14) μg/ml for collagen (n 10), 2·41 (se 0·11) μm for ADP and 385 (se 21) μm for arachidonic acid (n 10). Erythrocytes and leucocytes were added in a proportion to reach the cell counts in whole blood (4·8 (se 0·2) × 1012 erythrocytes/l and 6·0 (se 0·4) × 106 leucocytes/l).
Platelet production of thromboxane B2
The production of thromboxane A2 was determined by measuring its stable metabolite thromboxane B2. Samples of whole blood were stimulated with calcium ionophore A23187 (50 μm) at 37°C during 30 min, then they were centrifuged at 10 000 g for 3 min, and the amount of thromboxane B2 in the supernatant was determined with an enzyme immunoassay kit.
Leucocyte production of 6-keto-prostaglandin F1α
The production of prostacyclin was determined by measuring its stable metabolite 6-keto-PF1α. We used samples of leucocytes (6·5 (se 0·4) × 106 leucocytes/l) that were incubated for 5 min at 37°C, then 50 μm-calcium ionophore A23187 was added and the samples were shaken for 30 min. The samples were centrifuged at 10 000 g for 15 min at 4°C, and were frozen at − 80°C until analysis for 6-keto-PF1α by enzyme immunoassay.
Leucocyte production of nitric oxide
NO production was induced with the calcium ionophore A23187 (50 μm) and measured as nitrite+nitrate concentration in the supernatant of induced leucocyte samples (6·3 (se 0·3) × 106 leucocytes/l) with an enzyme immunoassay kit. The experiments were designed to determine NO production with the drug under study or in ethanol–PBS solution (control sample) for 10 min at 37°C. Briefly, leucocytes were incubated for 10 min at 37°C with 200 μl buffer containing 10 mm-HEPES, 1 mm-EDTA, 1 mm-dl-dithiothreitol (DTT), 10 μg/ml leupeptin, 4·8 mm-dl-valine, 1 mm-NADPH, 1 mm-MgCl2, 1 mm-CaCl2 and 20 μm-l-arginine, and then with 10 μl saline or the corresponding concentration of the drugs. Basal nitrite+nitrate production was first determined; after this step calcium ionophore A23187 was added and after 30 min of incubation at 37°C the increase in nitrite+nitrate production was evaluated.
3-Nitrotyrosine and TNFα production
Blood was incubated with bacterial membrane lipopolysaccharide (LPS; 1 μg/ml) for 18 h at 37°C. The plates were then centrifuged, and the plasma was removed and frozen at − 80°C. TNFα and 3-nitrotyrosine (an indicator of peroxynitrite production) were measured in the supernatant with a commercial enzyme immunoassay kit. In each experiment a sample of blood without LPS induction was incubated, and LPS-induced TNFα and 3-nitrotyrosine production was calculated as the difference between the LPS-induced samples and non-induced samples.
Statistical analysis
The data are expressed as means and their standard errors for ten experiments. All statistical analyses were done with the Statistical Program for Social Sciences version 12.0 (SPSS Inc., Chicago, IL, USA). One-way ANOVA followed by Bonferroni transformation and unpaired Student's t test were used, and differences were considered significant when P < 0·05.
Results
HT-AC and HT inhibited platelet aggregation induced by ADP, collagen or arachidonic acid in a concentration-dependent manner in whole blood and PRP (Figs. 1 and 2). Maximum intensity of aggregation in cultures induced with ADP was 23·51 (se 1·95) ohms in whole blood and 19·47 (se 1·10) ohms in PRP. After induction with collagen, maximum aggregation was 21·24 (se 1·16) ohms in whole blood and 18·97 (se 1·15) ohms in PRP, and in cultures induced with arachidonic acid, maximum aggregation was 23·49 (se 0·93) ohms in whole blood and 23·75 (se 0·95) ohms in PRP. Table 1 shows the concentrations of each compound that inhibited maximum platelet aggregation in control samples (incubated without the aggregation inducer) by 50 % (IC50). In whole blood, the antiplatelet aggregating effect of HT was significantly weaker than that of HT-AC or ASA. Inhibition of aggregation in whole blood was similar with HT-AC and ASA except when arachidonic acid was the inducer; in these experiments the antiaggregating effect of ASA was stronger. In samples of PRP induced with ADP or collagen we found no significant differences among the three compounds, but when arachidonic acid was the inducer, the effect of HT was significantly weaker than HT-AC or ASA.
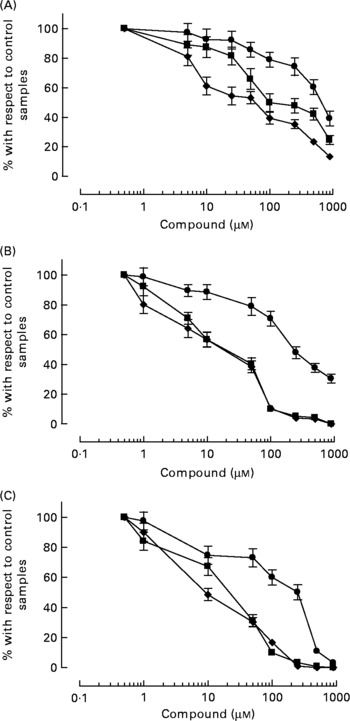
Fig. 1 Platelet aggregation in human whole blood after incubation with hydroxytyrosol acetate (■), hydroxytyrosol (●) and acetylsalicylic acid (♦). The inducers were ADP (2·5 μm; A), collagen (1 μg/ml; B) or arachidonic acid (400 μm; C). Values are means with their standard errors depicted by vertical bars.

Fig. 2 Platelet aggregation in human platelet-rich plasma after incubation with hydroxytyrosol acetate (■), hydroxytyrosol (●) and acetylsalicylic acid (♦). The inducers were ADP (2·5 μm; a), collagen (1 μg/ml; B) or arachidonic acid (400 μm; c). Values are means with their standard errors depicted by vertical bars.
Table 1 Concentrations of hydroxytyrosol acetate (HT-AC), hydroxytyrosol (HT) and acetylsalicylic acid (ASA) that inhibited 50 % of platelet aggregation (IC50), induced with 2·5 μm-ADP, 1 μg/ml collagen or 400 μm-arachidonic acid in whole-blood and platelet-rich plasma (PRP)‡
(Mean values with their standard errors)
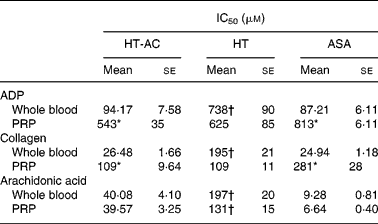
Mean values were significantly different from those of whole blood (unpaired Student's t test): *P < 0·0001.
Mean values were significantly different from those of ASA and HT-AC: †P < 0·0001.
‡ For details of subjects and procedures, see Experimental methods.
When we compared the antiaggregating effect in whole blood and PRP, we found that the effect of HT-AC and ASA was significantly stronger when aggregation was induced with ADP or collagen (Table 1); the antiaggregating effect of HT in whole blood was similar to that in PRP. In PRP incubated with leucocytes the antiaggregating effect of HT-AC and ASA increased in comparison to the effect in PRP alone (Fig. 3). In PRP incubated with erythrocytes the antiaggregating effects of HT, HT-AC and ASA were not different statistically with respect to PRP. Maximum aggregation was 21·75 (se 1·12) ohms in whole blood, 19·00 (se 0·56) ohms in PRP, 21·59 (se 1·74) ohms in PRP with erythrocytes and 16·08 (se 1·11) ohms in PRP with leucocytes.
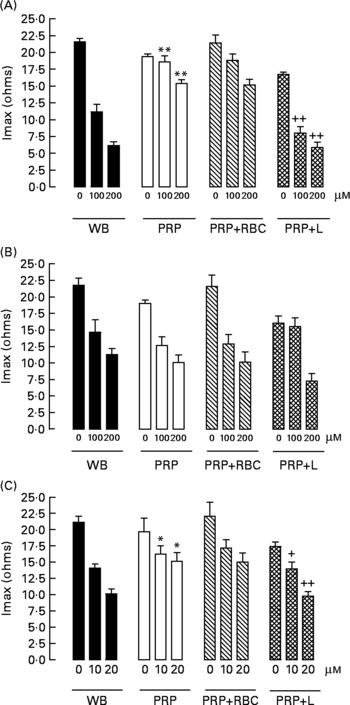
Fig. 3 Maximum intensity of platelet aggregation (Imax) induced with 1 μg/ml collagen in whole blood (WB), platelet-rich plasma (PRP), PRP plus erythrocytes (PRP+RBC) and PRP plus leucocytes (PRP+L) after incubation with hydroxytyrosol acetate (A), hydroxytyrosol (B) or acetylsalicylic acid (C). Values are means with their standard errors depicted by vertical bars. Mean values were significantly different from those of WB: *P < 0·05, **P < 0·001. Mean values were significantly different from those of PRP: +P < 0·05, ++P < 0·001.
All three compounds inhibited thromboxane B2 and 6-keto-PF1α production in a concentration-dependent manner (Fig. 4). Table 2 shows the IC50 values for these assays. The order of potency in the inhibition of these two prostanoids was ASA>HT-AC>HT. The thromboxane/prostacyclin inhibition ratio was 10·79 (se 1·03) for HT-AC, 1·05 (se 0·09) for HT and 3·28 (se 0·21) for ASA. Thromboxane B2 production in control samples was 1·29 (se 0·09) nmol/109 platelets and 6-keto-PF1α production was 1·16 (se 0·09) nmol/109 leucocytes.

Fig. 4 Whole-blood thromboxane B2 (A) and leucocyte keto-prostaglandin F1α (B) induced with 50 μm-calcium ionophore A23187 after incubation with hydroxytyrosol acetate (■), hydroxytyrosol (●) and acetylsalicylic acid (♦). Values are means with their standard errors depicted by vertical bars.
Table 2 Concentrations of hydroxytyrosol acetate (HT-AC), hydroxytyrosol (HT) and acetylsalicylic acid (ASA) that inhibited 50 % (IC50) calcium ionophore-induced thromboxane B2 and 6-keto-prostaglandin F1α (6-keto-PGF1α), lipopolysaccharide-induced 3-nitrotyrosine and TNFα, and concentrations of these compounds that increased calcium ionophore-induced nitrite+nitrate production by 50 % (EC50)‡
(Mean values with their standard errors)
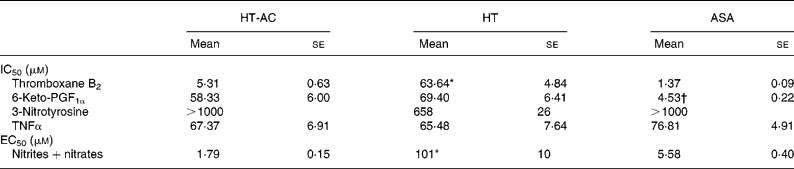
Mean values were significantly different from those of ASA and HT-AC: *P < 0·0001.
Mean values were significantly different from those of HT and HT-AC: †P < 0·0001.
‡ For details of subjects and procedures, see Experimental methods.
All three compounds increased nitrite+nitrate production in whole blood in a concentration-dependent manner (Fig. 5), although the effect of HT was significantly weaker than that of HT-AC and ASA (Table 2). Nitrite+nitrate concentration in control samples was 1·05 ± 0·01 μmol/l. The production of 3-nitrotyrosine induced with LPS was significantly inhibited only by the highest concentration of the compounds we tested (1000 μm) (Fig. 5; Table 2). The concentration of 3-nitrotyrosine in control samples was 39·85 (se 3·28) nmol/l. The production of TNFα was also inhibited in a concentration-dependent manner by all three compounds (Fig. 5), with no significant differences among them (Table 2). The concentration of TNFα in control samples was 10·36 (se 1·21) pg/ml.
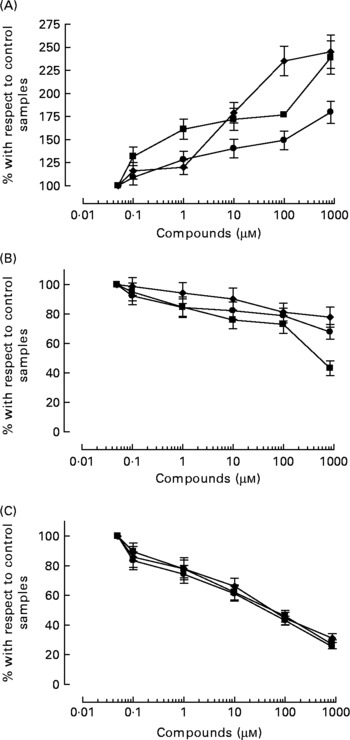
Fig. 5 Concentration of 50 μm-calcium ionophore-induced nitrites+nitrates in leucocytes (A), and 3-nitrotyrosine (B) and TNFα (C) induced with 1 μg/ml bacterial membrane lipopolysaccharide in whole blood, after incubation with hydroxytyrosol acetate (■), hydroxytyrosol (●) and acetylsalicylic acid (♦). Values are means with their standard errors depicted by vertical bars.
Discussion
We report the first evidence of the antiplatelet aggregating effect of HT-AC in human blood. The effect of HT-AC was stronger than that of HT and similar in quantitative terms to that of ASA. Some phenolic compounds in VOO inhibit platelet function, e.g., HT(Reference Petroni, Blasevich, Salami, Papini, Montedoro and Galli8, Reference Dell'Agli, Maschi, Galli, Fagnani, Dal Cero, Caruso and Boisio9), oleuropein(Reference Petroni, Blasevich, Salami, Papini, Montedoro and Galli8) and phenolic isochromans(Reference Togna, Togna, Franconi, Marra and Guiso13). In fact, feeding VOO to rats or rabbits was found to significantly reduce collagen-induced platelet aggregation(Reference De La Cruz, Villalobos, Carmona, Martín-Romero, Smith-Agreda and Sánchez de la Cuesta14, Reference González-Correa, Muñoz-Marín, Arrebola, Guerrero, Narbona, López-Villodres and De La Cruz15). The present results confirm this effect of HT, but also show that HT-AC has a greater antiaggregating effect than HT, and is almost as effective as ASA in blocking platelet aggregation. The latter observation is potentially important because ASA is the most widely used drug in the world in prophylaxis against thromboembolism.
In the present section we analyse the results obtained with HT and HT-AC in relation to the mechanisms of action for ASA, as the profile of inhibition of platelet aggregation of HT-AC is similar to that of ASA (Table 1; Figs. 1 and 2).
Firstly, the main mechanism that explains the antithrombotic effect of ASA is inhibiting the synthesis of thromboxane A2. All three compounds inhibit thromboxane synthesis in a concentration-dependent manner. It was previously shown that HT at a concentration of 400 μm in whole blood reduced collagen- or thrombin-induced thromboxane production by approximately 75 %(Reference Petroni, Blasevich, Salami, Papini, Montedoro and Galli8). Interpolating the results plotted in Fig. 4 for a concentration of 400 μm of the phenolic compounds compared here shows that HT would reduce thromboxane production by 67 % and HT-AC would reduce production by 86 %.
Secondly, the synthesis of thromboxane A2 can be reduced by inhibition of the enzyme cyclooxygenase (as acting ASA) or the enzyme thromboxane synthetase. The effect of HT and HT-AC cannot be attributed to the inhibition of thromboxane synthase activity, since prostacyclin production was also inhibited (Fig. 4). The present finding suggests that cyclooxygenase was probably the target of these phenols. More than 30 years ago this mechanism of action was shown in ASA(Reference Vane16), and it also appears to be important in the antiaggregating effect of HT-AC. Although platelet thromboxane and leucocyte prostacyclin production are not strictly comparable, they provide an approximate idea of the effect of these compounds on the thromboxane/prostacyclin balance through the IC50 prostacyclin/IC50 thromboxane ratio (see Results).
HT-AC inhibited thromboxane production more than prostacyclin production, an effect with clear benefits with a view to preventing thrombosis. The cyclooxygenase-inhibiting mechanism plays a potentially important role, since it was recently shown that HT has no effect on other pathways involving platelets such as cAMP, at least not at a concentration of 100 μm(Reference Dell'Agli, Maschi, Galli, Fagnani, Dal Cero, Caruso and Boisio9). The present findings are consistent with the weaker antiaggregating effect of both phenolic compounds and ASA when aggregation was induced by ADP, since the effect of ADP is mainly the result of its ability to lower cytoplasmic concentrations of cAMP(Reference De la Cruz, Bellido, Camara, Martos and Sanchez de la Cuesta17, Reference De la Cruz, Camara, Bellido, Carrasco and Sanchez de la Cuesta18). However, this is not to imply that inhibition of thromboxane synthesis is the only mechanism through which HT exerts its antithrombotic potential in vivo, since it has also been shown to inhibit the expression of adhesion molecules such as intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expressed on endothelial cells(Reference Dell'Agli, Fagnani and Mitro12).
Thirdly, other mechanisms can complete inhibition of cyclooxygenase produced by HT and HT-AC. This is supported in the differential antiaggregant effects in isolated platelets and in whole blood. We found that HT had the same antiaggregating effect in both media, whereas the effect of HT-AC and ASA was stronger in whole blood, albeit with some exceptions. When arachidonic acid was used as the inducer, aggregation resulted from the stimulation of platelet thromboxane A2 synthesis(Reference Zucker and Nachmias19). The absence of differences in the antiaggregant effect of ASA, HT and HT-AC is explained by the inhibition of cyclooxygenase. The greater antiaggregating effect of ASA and HT-AC in whole blood when platelets were induced with ADP or collagen might be due to an effect on a different biochemical pathway that mediates in the relations between leucocytes, erythrocytes and platelets. In this connection it was found that the greater effect of ASA in whole blood was mainly due to the stimulation of neutrophil NO production(Reference De la Cruz, Camara, Bellido, Carrasco and Sanchez de la Cuesta18, Reference Lopez Farre, Caramelo, Esteban, Alberola, Millas, Monton and Casado20), a finding also reported for other salicylate derivatives(Reference Sánchez de Miguel, Casado and Farré21).
Because erythrocytes did not modify the antiplatelet effect of HT and HT-AC, leucocytes could probably influence this effect through a modification in its NO production. ASA and the two phenolic compounds stimulated leucocyte NO production in a concentration-dependent manner (Fig. 5), although the effect of HT was smaller in quantitative terms than that of HT-AC or ASA. The reported ability of ASA to stimulate NO production was confirmed by the present finding that in PRP plus erythrocytes, the effect of ASA and HT-AC was similar to their effect in PRP alone, whereas in PRP plus leucocytes, the effect of HT-AC was similar to its effect in whole blood, as was also noted for ASA in an earlier report(Reference De la Cruz, Camara, Bellido, Carrasco and Sanchez de la Cuesta18) and in the present study (Fig. 3). HT has been shown to increase the cytoplasmic concentration of calcium and thus contribute to the increase in NO synthesis by endothelial cells(Reference Visioli, Bellosta and Galli22, Reference Visioli and Galli23). Our assays showed that HT-AC is also able to stimulate calcium-dependent NO production in human leucocytes.
Therefore, HT could inhibit platelet function in whole blood through an inhibition of platelet thromboxane A2 synthesis, while HT-AC could exert a greater antiaggregant effect because of the inhibition of thromboxane A2 synthesis plus an increase in NO leucocyte production, similar to the mechanism of action of ASA.
Fourthly, the effect of ASA on the production of inflammatory mediators has been related to its action in preventing CVD(Reference Shackelford, Alford, Xue, Thai, Adams and Pizzo24, Reference Mateos-Cáreres, García-Méndez and Farré25); accordingly, we tested TNFα production in leucocytes stimulated with LPS. All three compounds compared here inhibited the production of this inflammatory mediator in a concentration-dependent way, with no differences among them in the intensity of their effect. In this connection it has been shown that exposure of LPS-induced human THP-1 cells to olive vegetation water (containing approximately 200 μm-polyphenols) reduced TNFα production by 50 %(Reference Bitler, Viale, Damaj and Crea26). In the present experimental procedure, the effect of both HT and HT-AC was greater compared to the effect reported by Bitler et al. (Reference Bitler, Viale, Damaj and Crea26) Therefore, in addition to the antiplatelet effect of HT and HT-AC, it would be necessary to consider its effect on inhibiting the production of inflammatory mediators as an additional mechanism that could fill a possible effect in the prevention of CVD, similar to ASA.
Finally, in the genesis and evolution of vascular disease oxygen reactive species play an important role. In connection with the production of NO, the presence of free radicals facilitates peroxynitrite(Reference Beckman and Koppenol27), we wished to study the potential effect of free radicals on 3-nitrotyrosine formation as an indirect indicator of peroxynitrite production(Reference Tarpey, Wink and Grisham28). Despite the antioxidant effect of polyphenols in VOO(Reference Mateos, Dominguez, Espartero and Cert29, Reference Gordon, Paiva-Martins and Almeida30), these compounds failed to inhibit peroxynitrite production, at least under the present experimental conditions (Fig. 5). Moreover, this antioxidant effect occurs in a range of concentrations greater than the inhibition of platelet function, therefore it does not appear that the antioxidant effect of these compounds has a direct relationship with its antiplatelet effect. In addition, blood samples used for platelet aggregation do not have conditions for formation of free radicals, so the antioxidant effect of HT and HT-AC could not influence their antiplatelet effect.
The in vivo significance of the results presented here with regard to inhibition of platelet aggregation by VOO phenols, mainly HT and HT-AC, must be related to its pharmacokinetic characteristics. After ingestion of 40 ml VOO with a phenolic content of 366 mg/kg olive oil, maximal plasma concentration of HT reached 25·83 μg/l in man(Reference Miró-Casas, Covas, Farré, Fitò, Ortuño, Weinbrenner, Roset and De La Torre31), which represents approximately 130 μmol HT/l. However, around 95 % of HT is present in plasma in conjugate forms, suggesting an extensive first pass intestinal/hepatic metabolism of the ingested primary forms(Reference Fitò, De La Torre, Farré, Khymenetz, Marrugat and Covas32). According to these data only 1–2 μmol free HT/l is present in human plasma after ingestion of VOO. Platelet aggregation curves obtained in the present study show around 20–30 % inhibition, and the corresponding thromboxane B2 production was 30–40 % for HT-AC (data obtained by extrapolating the results at 1–2 μmol/l). The possible inhibition of platelet function in the presystemic circulation, as has been demonstrated for ASA(Reference FitzGerald, Lupinetti, Charman and Charman33), and the need to investigate the pharmacokinetic parameters of the administration of HT or HT-AC, remain to be established in order to postulate the importance of these results in the prevention of cardiovascular events.
In summary, the present results show that HT-AC, a polyphenol present in VOO, exerts an antiaggregating effect on platelets that is greater in quantitative terms than the effect of HT and similar to the effect of ASA. This effect involves a decrease in platelet thromboxane synthesis and an increase in leucocyte NO production. The present findings, together with the reported antioxidant effect of HT-AC and its ability to inhibit TNFα production, suggest the need for further studies of the effect of HT-AC on the suite of mechanisms that regulate thrombogenesis in man.
Acknowledgements
This study was partially supported by a grant from the Ministerio de Ciencia y Tecnología, Spain (AGL-04-7935-C03-02). None of authors have any conflict of interest. We thank A. Pino for his invaluable technical assistance and K. Shashok for translating parts of the manuscript into English. J. P. D. L. C. and J. A. G. C. designed the study and prepared the manuscript. J. A. L.-V., R. A., J. A. G. C. and J. P. D. L. C. contributed to the successful execution of the experimental work. J. L. E. and G. R.-G. contributed to the chemical preparation, synthesis and analysis of the polyphenols used in the study.







