



Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive, neurodegenerative disorder typically resulting from biallelic deletions of the survival motor neuron 1 (SMN1) gene. Patients demonstrate a loss of motor neurons resulting in progressive skeletal muscle atrophy and weakness. SMA carrier frequency is 1 in 50 resulting in a disease prevalence of approximately 1 in 10,000 live births Reference Verhaart, Robertson and Wilson1 making SMA the most common genetic cause of childhood death.
The contiguous SMN1 paralog, SMN2, encodes amino acids that are identical to SMN1; however, an exonic point mutation in a putative exonic splice enhancer leads to the exclusion of exon 7 in about 90% of SMN2 mRNA transcripts. As such, each SMN2 copy only produces about 10% of the functional SMN protein ordinarily produced from a single, functional SMN1 copy. Reference Burghes and Beattie2
Historically, SMA has been categorized into three clinical types based upon the onset of clinical symptoms and the maximum motor milestone achieved. SMA type I is characterized by symptom onset before 6 months of age and an inability to sit or stand independently. Reference Markowitz, Singh and Darras3 This severe infantile-onset form of SMA accounts for up to 60% of cases with a mean survival of 8–10-1/2 months of age. Reference Finkel, McDermott and Kaufmann4,Reference Kolb, Coffey and Yankey5 SMA type II shows symptom onset from 6 months to 18 months old with infants achieving the ability to sit but not walk independently. It has been estimated that 25% of children with SMA will have this form of the disease. SMA type III is characterized by symptom onset after 18 months old and children achieving the ability to walk at least some point during their lives. Reference Wadman, Wijngaarde and Stam6 Rarer forms of SMA can include a congenital form (type 0) or a late, adult-onset form (type IV) with each accounting for <1% of all cases of this disease. Reference Wadman, Wijngaarde and Stam6,Reference Juntas Morales, Pageot, Taieb and Camu7
SMN2 copy number has some predictive value with respect to the type of SMA. For example, approximately 80% of SMA type I infants have two SMN2 copies or less with a remainder having three SMN2 copies. Reference Calucho, Bernal and Alías8 Children diagnosed with SMA type II are most likely to have three SMN2 copies (80%) with approximately 16% having two SMN2 copies and 4% having four SMN2 copies. For SMA type III, 90% of children will have three or four SMN2 copies. Reference Calucho, Bernal and Alías8 Individuals with adult-onset SMA type IV have four or more SMN2 copies. Notwithstanding these associations, the SMN2 genotype–phenotype correlation is insufficient to allow unequivocal SMN2 genotype-based prediction of discrete SMA I, II, and III diagnoses.
Therapies have emerged for the treatment of SMA. In June 2017, nusinersen (Spinraza®) was approved by Health Canada and, at the time of writing, reimbursement criteria have been established in all but one Canadian province. An antisense oligonucleotide, nusinersen is administered at set intervals via intrathecal injection and binds to a segment of SMN2 pre-mRNA, altering its splicing and promoting the inclusion of exon 7. Reference Corey9 As a result, it augments the amount of full-length SMN protein produced enhancing survival of motor neurons in affected individuals with resulting significant clinical improvement. Nusinersen has been shown to be particularly effective when administered to SMA babies with two or three SMN2 copies before symptom onset. Reference De Vivo, Bertini and Swoboda10 Children with four SMN2 copies have not been studied in these pivotal clinical trials. Additional treatments are emerging for SMA with a beneficial response reported from preliminary clinical trials of onasemnogene abeparvovec (Zolgensma®), a gene replacement therapy Reference Mendell, Al-Zaidy and Shell11 as well as RO7034067 (Risdaplam®) a small molecule that also alters SMN2 splicing increasing full-length SMN2 mRNA production. Reference Sturm, Günther and Jaber12 These treatments have received regulatory approval in other jurisdictions and will require review by regulatory bodies in Canada.
Newborn Screening Ontario (NSO), based at CHEO in Ottawa, began operations in 2006. Prior to this, screening in the province of Ontario was based out of the provincial public health laboratories and screening targets were limited to phenylketonuria and congenital hypothyroidism. NSO has expanded the provincial NBS panel to now include over 25 conditions and the resulting identification and treatment of over 2500 infants affected by one of these conditions. As outlined in a seminal 1968 WHO publication by Wilson and Jungner, Reference Wilson and Jungner13 “the object of screening for disease is to discover those among the apparently well who are in fact suffering from disease”. Newborn screening (NBS) specifically is a public health population-based system, which involves testing all infants shortly after birth to identify those at risk for an increasing number of treatable conditions, which are not clinically evident in the newborn period. Reference Zavon14 Wilson and Jungner also delineated several fundamental principles of screening, which have been interpreted over time as “criteria” to be used in the consideration of the appropriateness of a given disease for inclusion in a screening program. These principles/criteria include characteristics of the disease itself (e.g. severity, knowledge of natural history), the screening test (e.g. test performance, robustness), the treatment (e.g. effectiveness, acceptability, and accessibility), and societal considerations (e.g. cost-effectiveness, harms to those with false-positive result including the risk of overtreatment). The emergence of access to nusinersen as a transformative therapy for SMA, along with the availability of robust and accurate DBS screening tests, the detailed understanding of the natural history of the disease, and the availability of a system of care for screen-positive infants involving pediatric academic health science centers has made SMA particularly appealing for inclusion in NBS programs. Potential concerns about the inclusion of SMA as a target of NBS include the high cost of treatment, challenges in predicting the severity of disease in infancy (with the associated risk of overtreatment of infants with less severe forms of SMA), and equitable and timely access to care across an area as large and, in places, as sparsely populated as Ontario. The NSO Advisory Council reviewed the evidence and recommended that SMA be added to the provincial NBS program. However, in order to maximize benefits and minimize potential harms of SMA NBS, and ensure consistency across the province, it was recognized that a strong consensus was needed on the approach to SMA screening, clinical evaluation, and treatment. Therefore, a group of Ontario screening and pediatric neuromuscular experts met prior to the initiation of pilot screening to define which individuals should be reported as positive (i.e. SMN1 deletion with or without consideration of SMN2 copy number), and for whom, among these, immediate treatment versus careful follow-up should be recommended. Shortly thereafter, on January 13, 2020, an SMA pilot program was initiated at NSO. The SMA test was multiplexed with a recently established hearing impairment and severe combined immunodeficiency mutation screening test, allowing a transition with relative ease, no additional blood sample requirements, and minimal additional testing cost.
Methods
Newborn Screening Testing Methodology
NBS dried blood samples (DBS) are collected on specially designed filter paper (also known as Guthrie paper) according to published criteria ideally from infants between 24 and 48 h of age. An insert outlining the SMA NBS pilot project was given to all parents with details on where to find further information on the NSO website.
The SMA NBS pilot in Ontario includes a laboratory-developed first-tier MassARRAY test for the presence of SMN1 and a second-tier multi-ligand probe amplification (MLPA) test for both SMN1 and SMN2 copy numbers (MRC Holland P021). The MassARRAY (Agena) test involves initial PCR amplification of the relevant SMN1 genomic region followed by annealing of primers overlapping or adjacent to sites of interest, with a single-base extension. SMA genotyping was added to a large multiplex MassARRAY assay assessing 22 mutations associated with early-onset hearing loss (GJB2/GJB6 and SLC26A4) as well as mutations within two genes causing severe combined immunodeficiency (SCID; IKBKB, and ZAP70) already being performed at NSO. Although many screening laboratories have multiplexed SMA PCR with TREC (SCID) qPCR, Reference Taylor, Lee and Yazdanpanah15 these tests had already been multiplexed with an assay for congenital CMV at NSO; additional targets in this qPCR assay were not possible. The first-tier screen performed at NSO, therefore, assesses for the presence of an SMN1 exon 7 single-nucleotide variant (SNV), and of exon 8 SMN1 and SMN2 SNVs. In traditional MassARRAY design, one primer would anneal to both SMN1 and SMN2, however, we noted during assay development that SMN2 copy number impacted SMN1 genotyping; specifically, individuals with ≥5 copies of SMN2 were often incorrectly genotyped as SMN1 null. To address this issue, we altered our design to independently assess SMN1 exon 7 by using a primer whose 3′ base is the C>T variant such that it will only anneal and produce a signal from SMN1. The exon 8 assay follows the traditional design where the primer sits adjacent to the exon 8 SNV and the signal is produced from both SMN1 and SMN2 (Figure 1). Given SMN1 and SMN2 dual null individuals are not viable, an exon 8 signal is anticipated in every live-born individual; if none is observed then a failed reaction is likely and the reaction is repeated. This method does not identify SMN1 +/− SMA carriers who would produce a signal indistinguishable from SMN1 +/+ individuals.
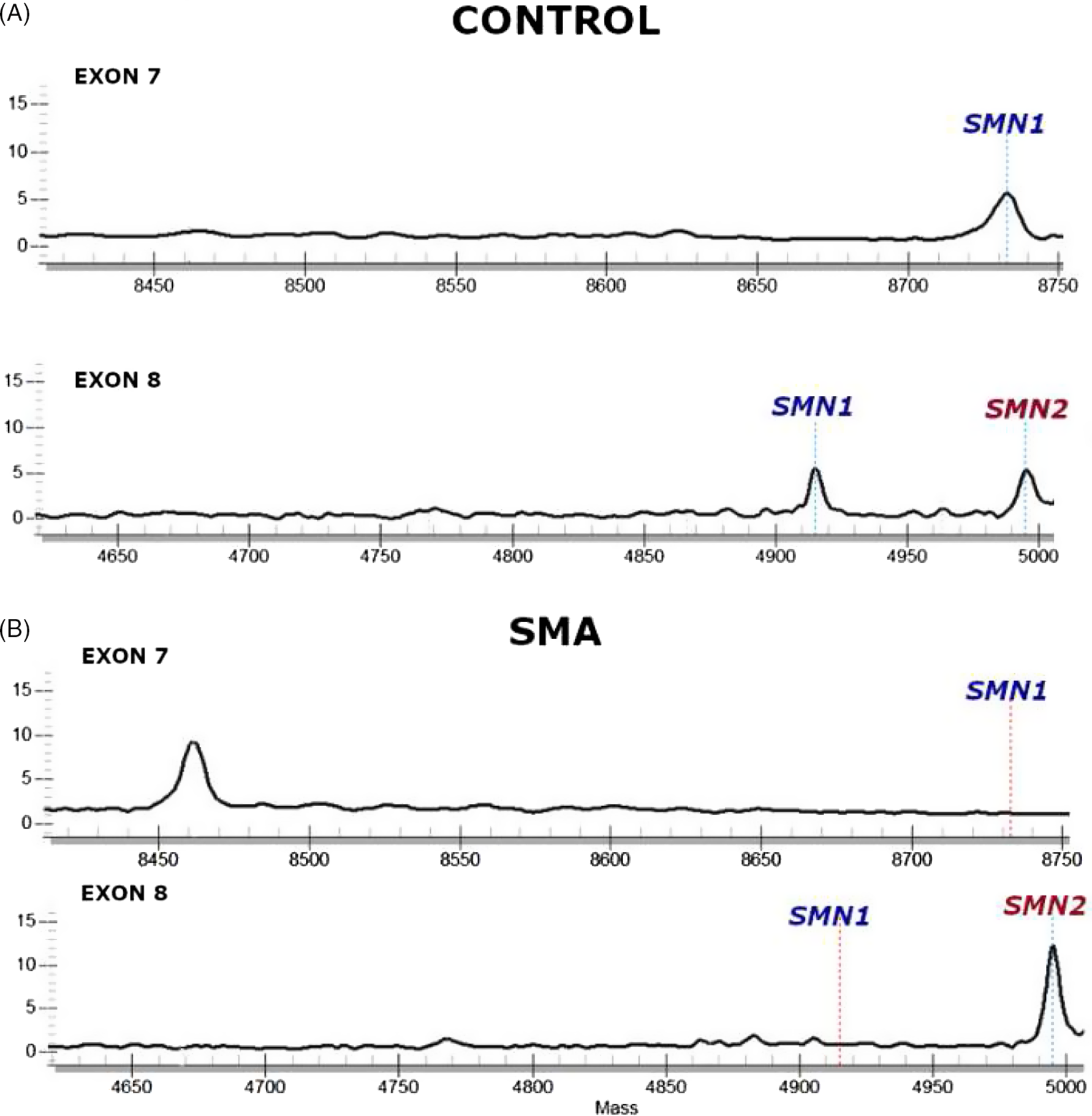
Figure 1: SMN1 and SMN2 MassARRAY chromatograms.
(A) Infants not affected with SMA (“control”) with one or two SMN1 copies will show a peak at SMN1 in both the exon 7 and exon 8 assays. (B) Infants affected with SMA (bottom) will not show SMN1 peaks in either exon 7 or exon 8 reflecting homozygous SMN1 deletion. The presence of SMN2 is seen just downstream (right) of where the SMN1 signal is seen (controls) or would have been expected (in affected infants), though this is not a target of the initial assay.
First-tier-positive samples (SMN1 null) are immediately analyzed by MLPA (MRC Holland P021) for SMN1 and SMN2 copy numbers and if confirmed, a screen-positive report which includes SMN2 copy number is issued if ≤4 x SMN2 copy numbers are detected. Samples with first-tier inconclusive results (exon 8 SMN1 null, exon 7 SMN1 present) are most likely due to high SMN2 copy number and thus analyzed by MLPA on a weekly basis.
Neuromuscular Disease Expert Consensus
In a series of teleconferences culminating in a 1-day face-to-face meeting, Ontario-based Pediatric Neuromuscular disease experts reviewed and discussed the evidence, expert consensus statements, provincial and national treatment reimbursement guidelines, and clinical practice regarding diagnosis and treatment of children with SMA as it pertained to NBS. The definition of a screen-positive result (specifically as related to SMN2 copy number) and details of a post-referral evaluation and management plan (including timelines) were then discussed and a post-referral evaluation algorithm was established.
Results
Newborn Screening Evaluation Algorithm
NSO medical staff (PC) in consultation with Ontario Pediatric Neuromuscular experts (CC, JD, HG, AM, HM, and MT) created a post-referral evaluation algorithm (Figure 2) comprising the following key points:
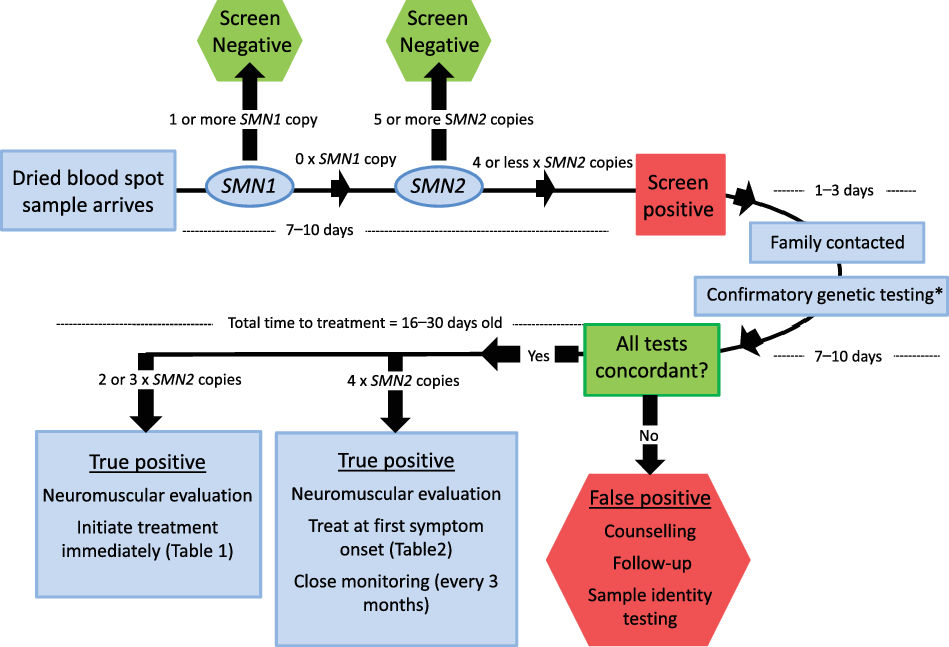
Figure 2: Post-referral evaluation algorithm.
All SMN1-null infants with four (or less) SMN2 copies will be reported. Patients’ families will be contacted by a genetic counselor or nurse within 3 days who will relay the test result and arrange for confirmatory genetic testing and an urgent meeting with a pediatric neuromuscular specialist. *Denotes the need for confirmatory multi-ligand probe amplification (MLPA) to be sent to a clinical laboratory (either SickKids Hospital or the Children’s Hospital of Eastern Ontario) as well as repeat sample to NSO laboratory. Patients with 2 or 3xSMN2 copies will receive immediate initiation of disease-modifying treatment. Patients with 4xSMN2 copies will be followed every 3 months by a pediatric neuromuscular specialist, with treatment initiation at the earliest sign of symptom onset. The goal for initiation of treatment is within the first 16–30 days of life.
It was decided that SMN1-null infants with four or fewer SMN2 copies would be classified “screen positive”. The group agreed that while the natural history of infants with 5xSMN2 copies or more was not wholly predictable, adult-onset disease or potentially remaining completely asymptomatic throughout his or her life was the most likely outcomes. As such, reporting this condition when there is a chance that disease manifestation may not occur was deemed to be unethical and not in patients’ best interest given the potential psychosocial impact, exclusion from insurability, and other potential ramifications associated with this disclosure.
The target time for the initial screen result was 7–10 days of age, acknowledging that samples may be taken at several days of age and/or shipped from remote sites within the province. All SMN1-null infants from the initial assay would undergo timely reflex MLPA testing, both confirming SMN1 −/− genotype and delineating SMN2 copy number.
SMN1 −/− infants with four SMN2 copies or less would be referred to a regional treatment center. A trained genetic counselor or nurse would contact the infant’s family by telephone and they would either be directed to the closest pediatric hospital or have blood sent for confirmatory SMA genetic testing to be performed and to meet with a pediatric neuromuscular specialist to discuss the potential implications of the NSO test result. One tube of whole blood (EDTA, 3 ml) would also be sent at the same time to the NSO laboratory for quality assurance purposes. Confirmatory testing would be performed at either SickKids Hospital (Toronto) or the Children’s Hospital of Eastern Ontario (Ottawa) aiming to return results within 7–10 days from receipt of the sample. One tube of whole blood (EDTA, 3 ml) would also be sent at the same time to the NSO laboratory for quality assurance purposes and rapid resolution of any discordant results (i.e. between the original screening results, diagnostic lab results, and second sample results at NSO). The rationale for the rapid investigation would be to determine if there was any evidence for the misidentification of specimens at the referring center or the receiving NBS laboratory to ensure there was no further delay in diagnosis and treatment initiation for another infant.
Following diagnostic confirmation, and determination of SMN2 copy number, infants, and their families are assessed by a pediatric neuromuscular specialist at which time the family would have an opportunity to discuss treatment options and standard of care guidelines that are followed at all Ontario Pediatric Neuromuscular clinics. Reference Mercuri, Finkel and Muntoni16,Reference Finkel, Mercuri and Meyer17 Baseline functional assessments (CHOP-INTEND, HINE) would be performed by a trained physiotherapist or clinical evaluator at or around that time. Establishing a definitive diagnosis between 16 and 27 days of age was determined to be feasible, and would allow initiation of disease-modifying therapy in the majority of SMA patients with 2 or 3xSMN2 copies by 30 days of life.
Treatment and Surveillance Recommendations: 2 or 3 SMN2 Copies
All infants with two or three SMN2 copies, given the evidence for rapid and irreversible loss of motor neurons, were recommended for immediate initiation of disease-modifying therapy prior to any clinical symptom onset (Figure 2). This recommendation is concordant with Ontario’s Exceptional Access Program (EAP) reimbursement criteria for nusinersen. 18 Application for access to nusinersen is to be placed with the family’s private insurance (if applicable) and/or the EAP. Neurophysiological testing (i.e. nerve conduction studies, electromyography (EMG)) was not recommended for children in this cohort as it would not alter treatment decisions. Although extremely uncommon, SMN1-null infants with only 1 SMN2 copy (i.e. predictive of SMA type 0) would be evaluated immediately. Given the potential severity of this congenital-onset form of SMA which could include the need for mechanical ventilation, the pediatric neuromuscular physician and family would discuss potential treatment options. Details of the recommended ongoing surveillance of patients with two or three copies of SMN2 are outlined in Table 1.
Table 1: Surveillance recommendations for infants with two or three SMN2 copies
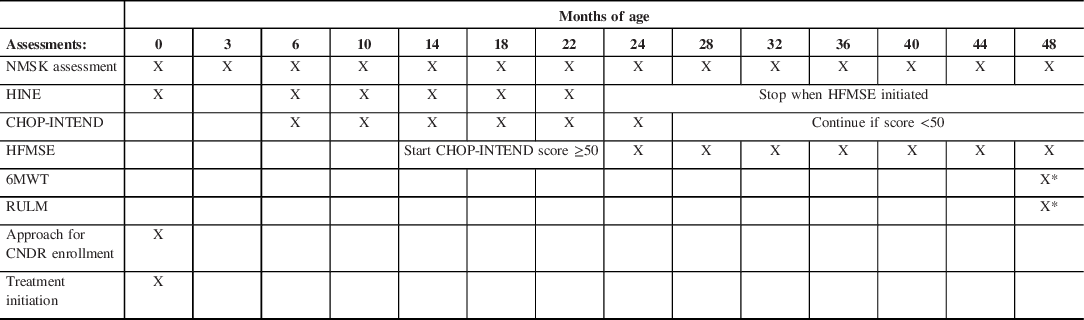
X denotes the age (in months) at which each assessment is performed. NMSK = pediatric neuromuscular assessment. Treatment will be initiated at the first visit and administered at 16–30 days of life. The Hammersmith Infant Neurological Examination (HINE) will be discontinued when the HFMSE = Hammersmith Functional Motor Scale Extended is initiated. CHOP-INTEND scoring will be discontinued at age 24 months old unless the score is <50 (maximum CHOP-INTEND score is 64). X* denotes that 6-min walk test (6MWT) and Revised Upper Limb Module (RULM) will be at 4 years of age if the child is developmentally capable of cooperating with this test.
Treatment and Surveillance Recommendations: 4x SMN2
Once confirmatory testing identifies four SMN2 copies in an SMA infant, an assessment is to be conducted by a Pediatric Neuromuscular expert since a proportion of infants with four SMN2 copies develop type I (<2%) or type II SMA (11%). Reference Calucho, Bernal and Alías8 Any clinical sign of SMA on neuromuscular examination (i.e. weakness, hypotonia, hyporeflexia, etc.) would prompt initiation of disease-modifying therapy. Motor nerve studies were recommended from the ulnar nerve (to abductor digiti minimi) and common peroneal nerve (to tibialis anterior). If the compound motor action potential (CMAP) was found to be <80% the lower-limit for age or if needle EMG noted any sign of denervation this would also confirm neurophysiological evidence of disease onset and prompt treatment initiation. If no clinical or neurophysiological evidence of disease was noted, it was recommended that treatment not be initiated and the child be seen every 3 months until 12 months of age. Surveillance physical examinations would be performed by a pediatric neuromuscular specialist and validated functional assessments (CHOP-INTEND, HINE) would be completed by a trained clinical evaluator until at least 2 years of age. Asymptomatic children with four SMN2 copies beyond 2 years of age are to be regularly assessed with the Hammersmith Functional Motor Scale Extended (HFMSE). The 6-min walk test (6MWT) and Revised Upper Limb Module (RULM) would be initiated at 4 years of age if the child was deemed developmentally capable of cooperating with this test. The complete recommended schedule of assessment is summarized in Table 2. Treatment would be recommended at the earliest clinical or electrophysiological sign of disease symptoms in this cohort.
Table 2: Surveillance recommendations for infants with four SMN2 copies
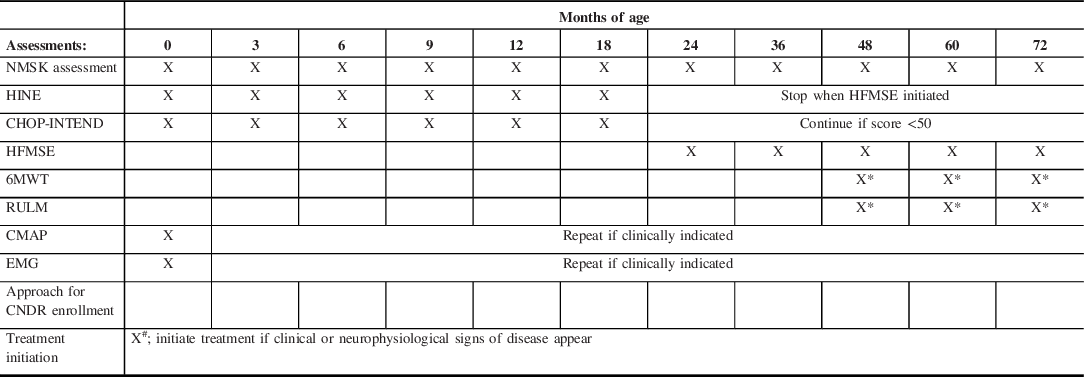
X denotes the age (in months) at which each assessment is performed. NMSK = pediatric neuromuscular assessment. Infants with 4xSMN2 copies who show clinical or neurophysiological signs of disease onset (i.e. compound muscle action potential (CMAP) amplitudes <80% lower limit of normal and/or needle electromyography (EMG) demonstrating an evidence of denervation) will prompt initiation of treatment (denoted by X#). Declining motor function scores will prompt initiation of treatment. Infants not showing evidence of disease onset will have a repeat examination and motor function scoring every 3 months (until 12 months old) where after they will be seen at 18 months old, 24 months old, and then annually. The Hammersmith Infant Neurological Examination (HINE) will be performed every 3 months until the HFMSE = Hammersmith Functional Motor Scale Extended is initiated. CHOP-INTEND scoring will be discontinued at age 24 months old unless the score is <50 (maximum CHOP-INTEND score is 64). X* denotes that 6-min walk test (6MWT) and Revised Upper Limb Module (RULM) will be performed beginning at 4 years of age if the child is developmentally capable of cooperating with this test. CMAP or EMG will be repeated only if needed to guide decision-making regarding treatment initiation.
Limitations of Testing
As with all other SMA NBS approaches currently in use, our screening platform will only identify patients with an SMN1 gene deletion or conversion, but not those with other pathogenic variations in SMN1 such as point mutations or small deletions. This is predicted to miss 3–5% of children who may have point mutations on one or both alleles. As such, a level of clinical vigilance must be maintained when seeing screen-negative infants and children with an SMA phenotype including, when indicated, neurophysiological testing (i.e. EMG) and/or confirmatory Sanger sequencing.
Discussion
The remarkable progress in SMA therapies, along with advances in NBS technology, has made the disease an excellent candidate for inclusion in screening programs. It is clear that early initiation of treatment, ideally in pre-symptomatic or minimally symptomatic SMA infants offers the best chance of optimizing motor function, reducing complications such as respiratory insufficiency requiring ventilation as well as indefinitely prolonging survival. Reference De Vivo, Bertini and Swoboda10 In keeping with this, many countries and jurisdictions have adopted NBS for SMA. Following the inclusion of SMA in the American Recommended Uniform Screening Panel (RUSP) in July 2018, 37 individual states have adopted NBS for SMA with 23 of those states having implemented full screening programs as of May 2020. 19 An additional three states have undertaken pilot screening programs. 19
The ability to predict natural history in asymptomatic patients diagnosed during screening is vital to make rational treatment decisions and appropriately counsel families. While the overlap in observed disease severity between those with various SMN2 copy numbers means it is an imperfect predictor for this purpose, it is nevertheless an important parameter in guiding such decisions. We recommend treating all babies with two or three SMN2 copies, in alignment with provincial drug reimbursement guidelines. The recommended deferral of treatment of infants with four SMN2 copies pending clinical or electrophysiological evidence of disease onset follows provincial drug reimbursement guidelines as well as the approach used in a recent German pilot Reference Vill, Kölbel and Schwartz20,Reference Müller-Felber, Vill and Schwartz21 and previously published American treatment guidelines, Reference Glascock, Sampson and Haidet-Phillips22 but diverges from a revision of the latter guideline which recommends early treatment. Reference Glascock, Sampson and Connolly23 More recently, an NBS pilot program in New South Wales and the Australian Capital Territory (NSW/ACT) did not report patients with four or more SMN2 copy patients. Reference Kariyawasam, Russell and Wiley24 In this regard, our recommendation to not treat all four SMN2 copy SMA patients was grounded in the observation that there is heterogeneity with regard to the age of symptom onset. Although patients may show symptom onset at around 3 years of age, a subset may have much later onset, not occurring until mid-adulthood (mean 37 years old; range 30–43 years old). Reference Wadman, Stam and Gizen25 This as well as the paucity of clear evidence for the efficacy of treatment in the four SMN2 copy groups was the basis for the decision for careful surveillance in this cohort. It is also very important to note that this decision was also guided by core screening principles including access to treatment within the system of care, avoidance of harm from overtreatment, and cost-effectiveness considerations.
The rapid initiation of treatment is essential, particularly for pre-symptomatic, 2xSMN2 copy patients who show a rapid decline in neurophysiological markers (e.g. CMAP amplitudes) shortly after birth. Reference Swoboda, Prior and Scott26 Over 40% of SMA patients identified in an NBS program show clear evidence of clinical symptoms within the first few weeks of life. Reference Kariyawasam, Russell and Wiley24 We have established a target range of 16–30 days of age (Figure 2) within which to initiate treatment, similar to the NSW/ACT program which started disease-modifying treatment at a median of 26.5 days of life (range: 16–37 days). Reference Kariyawasam, Russell and Wiley24 Optimally, by decreasing the time for confirmatory genetic testing and/or reducing time for EAP treatment approval, we hope to initiate treatment before 21 days of life. Currently, EAP approval cannot proceed until confirmatory genetic testing has been obtained. The demonstration of a strong concordance between NBS and confirmatory genetic test results could potentially allow a process of conditional EAP approval pending confirmatory testing results.
The profound alteration of SMA natural history observed following early treatment alters the historical concept of SMA “typing” based upon the age of symptom onset and the highest motor milestone achieved. There is strong evidence that initiation of therapy in the pre- or early symptomatic phases of disease is vital to optimizing outcomes for children with early-onset SMA. Reference De Vivo, Bertini and Swoboda10,Reference Mendell, Al-Zaidy and Shell11 With NBS enabling early diagnoses and access to highly effective therapies, children with SMA should survive and achieve motor milestones that would not have previously been possible. While SMN2 copy number has been used as a surrogate for SMA type, the overlap between copy number and SMA type means that the evaluation of the effectiveness of treatment in individual patients cannot solely be based on copy number alone as it lacks sensitivity and specificity. Other genetic modifiers alter the severity of the SMA phenotype including the rare SMN2 c.859G>C mutation, which is associated with higher SMN2 protein levels. Reference Vezain, Saugier-Veber and Goina27 Actin-binding protein plastin 3 (PLS3), neurocalcin delta (NCALD), and neuronal apoptosis inhibitory protein (NAIP) have been reported to have possible disease-modifying effects. Reference Yanyan, Yujin and Jinli28–Reference Qu, Ge and Bai30 Given that there is currently insufficient evidence pertaining to the population prevalence, strength of association, and effect upon the phenotype for these genetic modifiers, testing was not incorporated into the screening algorithm. As evidence emerges, it will be necessary to consider including such testing, which may be particularly valuable for individuals with 4xSMN2 copies since they have the potential for considerable phenotypic heterogeneity. Additional means to predict natural history of disease will be needed both to guide individual patient treatment decisions, as well as for a better understanding of the effectiveness of NBS for SMA in changing outcomes.
Ongoing assessment of this program will be necessary, as will review of the current algorithms as further evidence, drug reimbursement guidelines, and clinical consensus statements emerge. Ultimately, as a better understanding of “4xSMN2 copy” SMA disease onset emerges from the NBS-based postnatal ascertainment of affected infants, a better sense of treatment timing will also be delineated. The high levels of SMN observed in both control fetal and postnatal spinal cord tissue drop dramatically so that by 3 months of age it is indistinguishable from levels seen in SMA postmortem tissue. Reference Ramos, d’Ydewalle and Gabbeta31 Thus, judiciously timed reduced doses in the first 6 months may afford long-term benefit, forestalling, and possibly even preventing disease onset for SMA patients with four copies of SMN2. In keeping with this, the elegant study of inducible SMA mouse models has shown minimal SMN requirement following maturation of the neuromuscular junction. Reference Kariya, Obis and Garone32 In addition, new biomarkers are under investigation which may enable further precision related to symptom onset and need for and response to therapy.
In the end, clinical trials of such treatment approaches in the subset of SMA patients mostly at risk for late-onset disease will be needed to support individual patient treatment decisions, drug reimbursement policies, and screening approaches and policies. As multiple disease-modifying therapies emerge, there will be a need to evaluate the efficacy and tolerability of treatments at different stages of the disease. As Wilson and Jungner elegantly described in their treatise, and as illustrated in our recommended approach, screening decisions and programs must always balance individual and population benefits and harms, taking into account disease, treatment, test, and social considerations. Ultimately, our hope is that this can provide useful information for Ontario physicians regarding the current landscape of NBS in the province as well as for other provinces that may be making decisions about screening programs of their own.
Acknowledgments
Biogen provided funding to AMK (Principal Investigator) to allow Newborn Screening Ontario (NSO) to carry out this pilot study.
Disclosures
KDK, EY, KA, JM, MAT, JV, and PC report no disclosures relevant to the manuscript. HJM has been a member of an AveXis Advisory Board and was a site investigator for an SMA clinical trial (AveXis). JB has been a member of Advisory Boards (Biogen, Novartis). CC has been a site investigator for SMA clinical trials (Biogen, Roche). JJD has been a consultant for a company developing therapies for rare diseases. HG has been a member of Advisory Boards (Biogen, Roche). AMK was the principal investigator and received research funding for the pilot newborn screening project (Biogen).
Authors Contributions
HJM, KDK drafted the manuscript. AMK, PC assisted with the writing of the manuscript. EY, KA, JJD, CC, JD, HG, JM, MAT, JV, AMK, and PC provided critical review and edited the manuscript. All authors approve of the manuscript in its current form.
Abbreviations
CHEO = Children’s Hospital of Eastern Ontario;
CHOP-INTEND = Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders;
CMAP = compound motor action potential;
CMV = cytomegalovirus;
DBS = dried blood spot;
HFMSE = Hammersmith Functional Motor Scale Extended;
HINE = Hammersmith Infant Neurological Examination;
mRNA = messenger ribonucleic acid;
MLPA = multi-ligand probe amplification;
NAIP = neuronal apoptosis inhibitory protein;
NBS = Newborn Screening;
NSO = Newborn Screening Ontario;
NSW/ACT = New South Wales and Australian Capital Territory;
PCR = polymerase chain reaction;
SCID = severe combined immunodeficiency;
SMA = spinal muscular atrophy;
SMN1 = survival motor neuron 1;
SMN2 = survival motor neuron 2;
SNV = single-nucleotide variant;
WHO = World Health Organization.




