



Amyotrophic lateral sclerosis (ALS) is a devastating rapidly progressive motor neuron disease with an incidence rate of 2 per 100,000 per year.Reference Wolfson, Kilborn, Oskoui and Genge 1 The Canadian health care system is administered and funded independently by each respective province, resulting in variations in accessibility to and quality of specialized care. Amyotrophic lateral sclerosis has a tremendous psychological and economic impact on Canadian patients and their caregivers. It has been demonstrated that centralized multidisciplinary ALS clinic care results in improved survival compared with community-based care.Reference Rooney, Byrne and Heverin 2 , Reference Traynor, Alexander, Corr, Frost and Hardiman 3 In Canada, there is a mean annual direct out-of-pocket cost of C$19,574 per patient with a mean annual income loss of C$36,467 per patient per year and C$20,353 per caregiver per year.Reference Gladman and Zinman 4 , Reference Gladman, Dharamshi and Zinman 5
The exact cause of sporadic ALS remains unclear, and there is no effective cure. However, riluzole, a glutamate antagonist that reduces glutamate-mediated excitotoxicity, modestly slows disease progression and extends median survival by 2-3 months.Reference Bensimon, Lacomblez and Meininger 6 , Reference Miller, Mitchell and Moore 7 However, five studies using large databases spanning 5-10 years have suggested that treatment with riluzole might be associated with a prolonged survival of 6, 10, 12, 14 or even 21 months. These cohort studies had longer-term follow-up than the clinical trials, but are subject to greater bias.Reference Miller, Jackson and Kasasrskis 8 Riluzole use has minimal adverse effects, and is prescribed to ~60% of patients in both Europe and North America.Reference Traynor, Alexander, Corr, Frost and Hardiman 3 Clinical care practice guidelines of the American Academy of Neurology recommend that patients should be offered riluzole to slow disease progression.Reference Miller, Jackson and Kasasrskis 8 The cost of riluzole in Canada ranges between C$400 and C$600 per month and coverage for riluzole is available through various insurance plans. Prescribing rules for riluzole vary among and within provinces, and it may only be prescribed by a neurologist or physiatrist in Canada. During the time of data collection, all seven provinces in the study included riluzole on the provincial formulary, and provided additional programs to help cover riluzole costs for those without private insurance. Saskatchewan included riluzole on the provincial formulary during data collection (in 2017). Canadian residents with financial need can be eligible to receive riluzole coverage through the Medicum Rilutek Reimbursement and Co-Pay Assistance program. Yet, patients’ knowledge and/or use of this privately sponsored coverage may vary province-to-province. Edaravone, an antioxidant with unknown mechanism of action, shown to modestly improve survival in early stage ALS, was not available in Canada during the time of data collection.Reference Abe, Aoki and Tsuji 9
Although there is currently no known cure for ALS, comprehensive symptomatic management is available. A shortened time to diagnosis allows for timely provision of prognostic information, counseling and institution of a clinical management plan including symptom management (i.e., spasticity, sialorrhea, pain, percutaneous endoscopic gastrostomy [PEG] tube and non-invasive ventilation). In addition, shorter time to diagnosis results in improved recruitment into clinical trials.Reference Zoccolella, Beghi and Palagano 10 - Reference Testa, Lovati, Ferrarini, Salmoiraghi and Filippini 12 Individuals with a shorter time to diagnosis are more likely to meet criteria for trial participation such as time from symptom onset less than 24 months or percent predicted forced vital capacity above 80%. Thus, earlier diagnosis would lead to a larger population of patients who are likely to better respond to life-prolonging therapy such as riluzole or potentially edaravone.Reference Abe, Aoki and Tsuji 9 , Reference Swash 11 - Reference Turner, Parton, Shaw, Leigh and Al-Chalabi 13
Similarly, a timely and accurate diagnosis can alleviate the anxiety associated with diagnostic uncertainty and enable patient and family planning.Reference Househam and Swash 14 The reasons that often contribute to a slower time to diagnosis include the following:Reference Belsh 15 - Reference Chio 17
∙ physician’s lack of familiarity with ALS;
∙ patient comorbidities;
∙ complexity of the referral system;
∙ difficulty of early clinical diagnosis when ALS may be restricted to one region of the nervous system;
∙ hesitancy by a health care provider in communicating the diagnosis of a progressively disabling and fatal disease.
Shorter time to diagnosis also prevents unnecessary investigations and treatments that could result in avoidable complications and health care expenditures. Studies have found that ~40% of ALS patients receive one or more misdiagnoses before a correct diagnosis of ALS. Further investigation showed that a subset of these misdiagnosed patients underwent unnecessary surgery for symptoms later attributable to ALS, resulting in increased health care expenditures and risks of complications.Reference Kraemer, Buerger and Berlit 18 - Reference Belsh 20 Therefore, time from symptom onset to diagnosis is an important measure that can have significant impact on the ALS journey for patients.
Symptom management in ALS includes nutritional management, as both malnutrition and body weight are independent prognostic factors for survival.Reference Stambler, Charatan and Cedarbaum 21 - Reference Marin, Desport and Kajeu 24 Dysphagia impairs swallowing and occurs secondary to bulbar involvement in ALS. This can be compounded by limb weakness affecting the ability to prepare and feed oneself. Without adequate nutrition, patients will become malnourished, leading to respiratory distress and reduced quality of life.Reference Heffernan, Jenkinson and Holmes 25
Some studies have demonstrated that the use of a feeding tube can improve survival; however, there is some debate regarding this, as well as the optimal timing of PEG tube intervention.Reference Thornton, Fotheringham, Alexander, Hardiman, McGrath and Lee 26 - Reference Mathus-Vliegen, Louwerse, Merkus, Tytgat and Vianney-de-Jong 29 However, it is clear that the use of a PEG tube can improve body weight and subsequent quality of life.Reference Chio, Finocchiaro, Meineri, Bottacchi and Schiffer 30 , Reference Silani, Kasarskis and Yanagisawa 31 On the basis of these findings, the AAN clinical care guidelines for ALS recommend placing of a PEG tube to supplement nutrition.Reference Miller, Jackson and Kasasrskis 8
Patient registries, such as the Canadian Neuromuscular Disease Registry (CNDR), are an important tool for health care planning through the collection of real-world patient data enabling comparative analyses among different countries and regions.
The purpose of this study was to assess provincial differences in care delivery, by assessing time to diagnosis and interventions, including riluzole, ventilation and feeding tube use, for ALS patients across Canada.
Methods
The CNDR collects prospective clinical data at ALS clinics in seven of ten Canadian Provinces and zero of three Canadian territories (Supplementary Table 1). Data set elements were derived by the consensus of a disease working group encompassing expert clinicians, geneticists and scientists from across Canada, as previously published.Reference Korngut, Genge and Johnston 32
The CNDR is administered through a national office with affiliated multidisciplinary neuromuscular and ALS clinics throughout Canada. Patients are required to have a diagnosis of ALS according to World Federation of Neurology EI Escorial—Revised criteria,Reference Brooks, Miller, Swash and Munsat 33 and must provide informed consent in order to be entered into the registry. Patient recruitment to the registry is ongoing in affiliated ALS clinics and through the national office. In Canada, patients are first referred from a primary care practitioner to a specialist (general neurologist, neuromuscular specialist or other) for diagnosis by electromyography, and then re-referred to a neuromuscular specialist and/or ALS specialty clinic (Figure 1). All patients analyzed in this study were seen in hospital-based multidisciplinary clinics.
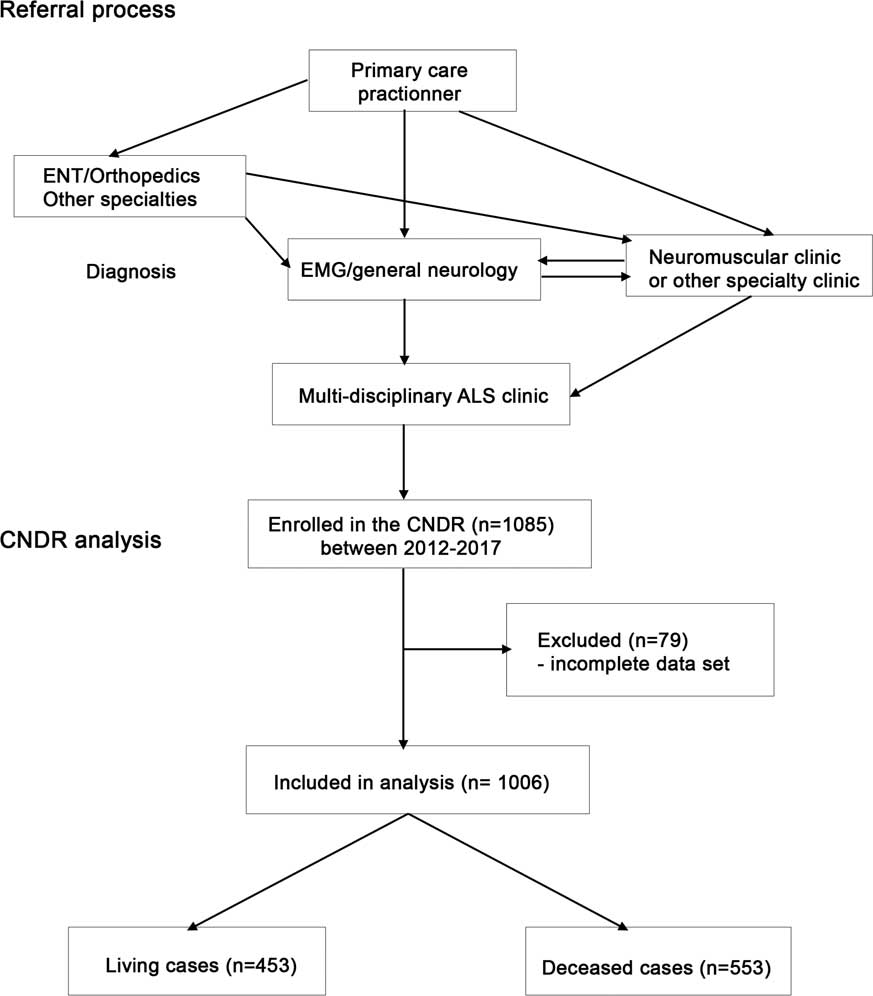
Figure 1 Diagram of Canadian Neuromuscular Disease Registry (CNDR) amyotrophic lateral sclerosis (ALS) patient flow and analysis. EMG=electromyography; ENT=ear, nose and throat.
Patient data are collected prospectively at routine clinic visits by the attending physician and trained data entry staff taking information from medical charts. Recruitment and data collection across different clinics is expected to be highly comparable owing to rigorous research assistant training, the availability of a comprehensive data dictionary defining each data item in detail and regular CNDR project manager teleconferences with data entry staff. Data integrity is ensured through auditing at the National Office.
Data collected are itemized in Supplementary Figure 1 and include the following items:
∙ date of diagnosis;
∙ date of symptom onset;
∙ riluzole use;
∙ feeding tube use;
∙ use of ventilation;
∙ amyotrophic lateral sclerosis revised functional rating scale (ALSFRS-R, a questionnaire-based disability scale);
∙ genetic testing.
Data were collected between 2010 and 2017 from both prevalent and incident cases. All data are collected in compliance with local research ethics board approvals. The following parameters were analyzed in this study:
∙ time to diagnosis (from first symptom onset);
∙ disease progression (ALSFRS-R progression rates, calculated as a decrease in ALSFRS-R scores divided by time between assessments);
∙ riluzole use at any point after the diagnosis;
∙ feeding tube use at any point after the diagnosis;
∙ ventilation use (either non-invasive or invasive) at any point after the diagnosis;
∙ survival (from first symptom onset).
Statistics were calculated using IBM SPSS Statistics for Macintosh, Version 24, with p<0.05 considered significant. Descriptive statistics were calculated from the patient’s first recorded clinic visit. Descriptive statistics are described as mean ± standard deviation. Mean descriptive statistics were compared with reported United States means using a paired one-way Student’s t-test.
Time from symptom onset to diagnosis was calculated for each patient, and means for each province were compared using a one-way analysis of variance (ANOVA), followed by a post-hoc pairwise Tukey’s Honest Significant Difference (HSD).
Riluzole use is recorded on the CNDR ALS physician form as one of the following: yes, no, past, stopped, declined or unknown. Riluzole use was calculated as “yes” if patients had recorded present usage, usage in the past or whether they stopped during any clinic visit. Riluzole use per province was compared using a χ 2 test.
Survival analysis was calculated as the amount of time in months from symptom onset to death. Cases were excluded for absence of date of death. Survival analysis per province was compared using a Log Rank χ 2 test, and median survival presented with a 95% confidence interval (CI). This is derived from log transformation of the survival (Kaplan-Meier) function.
Mean ALSFRS-R progression rates were calculated as mean difference per month, using a total score of 48 at time of symptom onset as a baseline value and calculated to the first recorded clinic visit. A score of 48 is considered normal and a score of 0 is considered severely impaired, with lower scores indicating increased impairment. Progression rates, assumed to be on a continuous scale, were compared using a one-way ANOVA with post-hoc, pairwise Tukey’s HSD.
Feeding tube use was recorded as “yes” if the patient had used a feeding tube at any point during the course of disease. Feeding tube use per province was compared using a χ 2 test.
Ventilation use was recorded as “yes” if the patient had ever used either non-invasive or invasive ventilation at any point during the course of disease. Ventilation use per province was compared using a χ 2 test.
An estimate of the number of living ALS patients in Canada at this time was calculated using the upper limit of international prevalence rates of 10 in 100,000 adults and a Statistics Canada 2016 Canadian population at risk estimate of 28,388,100 of adults older than 20 years. 34
Results
A total of 1085 ALS patients were registered in the CNDR at the time of analysis through participating clinics (Supplementary Table 1). After excluding individuals with incomplete data in the registry because of absence of a recorded date for symptom onset, absence of date of birth or lack of data regarding riluzole use, 1006 patients remained for analysis (Table 1, Figure 1). Using international prevalence data and Canadian population figures, we estimate that there are currently ~2800 living ALS patients in Canada. We report data on our cohort of 1006 patients (453 living patients). Mean age at onset (defined as first symptoms) was 60.1 (±12.0) years and mean age at diagnosis was 61.8 (±11.9) years. Median survival from onset was 36.5 months (95% CI 33.6-39.3). The population analyzed had more males than females, with males representing 60%. These statistics are comparable to available published data for survival (36 months), age at diagnosis and gender prevalence in the United States.Reference Horton, Graham and Punjani 35 , Reference Mehta, Kaye and Bryan 36
Table 1 Descriptive statistics for amyotrophic lateral sclerosis patient population in Canadian Neuromuscular Disease Registry (CNDR)

Mean time of symptom onset to diagnosis was significantly different among provinces (F=3.395, p=0.003). It was longest in Saskatchewan (27.0 months) and shortest in Nova Scotia (15.1 months). A comparison of inter-province differences is represented in Table 2. There was no significant difference among time to diagnosis across provinces for males compared with females (F=1.295, p=0.255; data not shown).
Table 2 Mean comparison between provinces for time between symptom onset and diagnosis
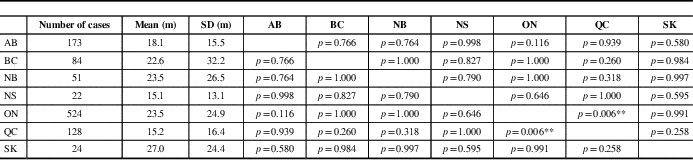
AB=Alberta; BC=British Columbia; m=months; NB=New Brunswick; NS=Nova Scotia; ON=Ontario; QC=Quebec; SK=Saskatchewan.
p values are represented in provincial comparisons.
**p<0.01 by Tukey’s Honest Significant Difference test (F=3.395, p=0.006).
Riluzole usage was also significantly different among provinces (p=0.000; χ 2=151.44), with the lowest usage in British Columbia (18.1%) and highest in Quebec (79.7%) (Figure 2). The national average (68%) was close to the expected usage rate of 60 % globally.Reference Traynor, Alexander, Corr, Frost and Hardiman 3
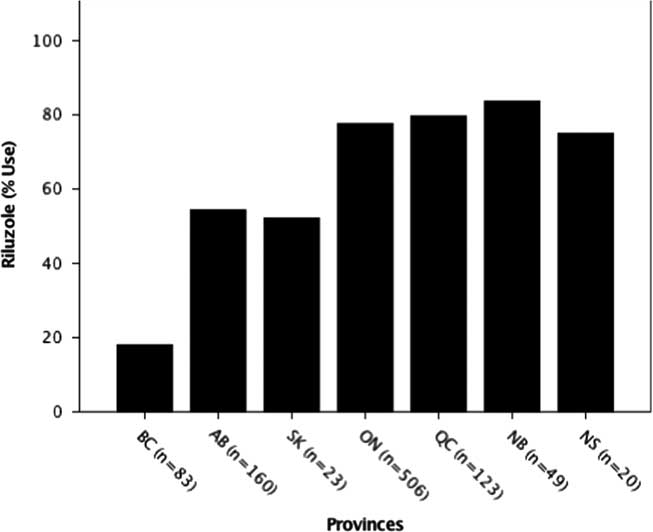
Figure 2 Riluzole use by province. The percent usage of riluzole was determined for each province. Usage by province was significantly different at ***p<0.001 (p=0.00; χ 2=151.44). AB=Alberta; BC=British Columbia; NB=New Brunswick; NS=Nova Scotia; ON=Ontario; QC=Quebec; SK=Saskatchewan.
Feeding tube use was significantly different across provinces (p=0.000; χ 2=35.54) (Figure 3), with the lowest usage in British Columbia (16.0%) and highest in Nova Scotia (52.6%).

Figure 3 Feeding tube use by province. The percent usage of feeding tubes was determined for each province. Usage by province was significantly different at ***p<0.001 (p=0.00; χ 2=35.54). AB=Alberta; BC=British Columbia; NB=New Brunswick; NS=Nova Scotia; ON=Ontario; QC=Quebec; SK=Saskatchewan.
Ventilation use was not significantly different across provinces; the mean usage across Canada was 31.7% (p=0.437; χ 2=5.88) (data not shown).
Mean ALSFRS-R progression rates in our cohort (0.75 units per month) are slightly slower than the reported mean progression rate of 0.9 units/month.Reference Takei, Tsuda, Takahashi, Hirai and Palumbo 37 , Reference Chio 38 Importantly, there was no significant difference (F=1.672, p=0.125) in disease progression rates across provinces as measured by ALSFRS-R scores (data not shown).
Despite the differences in time to diagnosis, riluzole use and feeding tube use, median survival of ALS patients (370 deceased patients) in Canada was 36 months (95% CI 33.6-39.3) from symptom onset and did not demonstrate any significant differences among provinces (p=0.167, χ 2=9.113) (Figure 4).
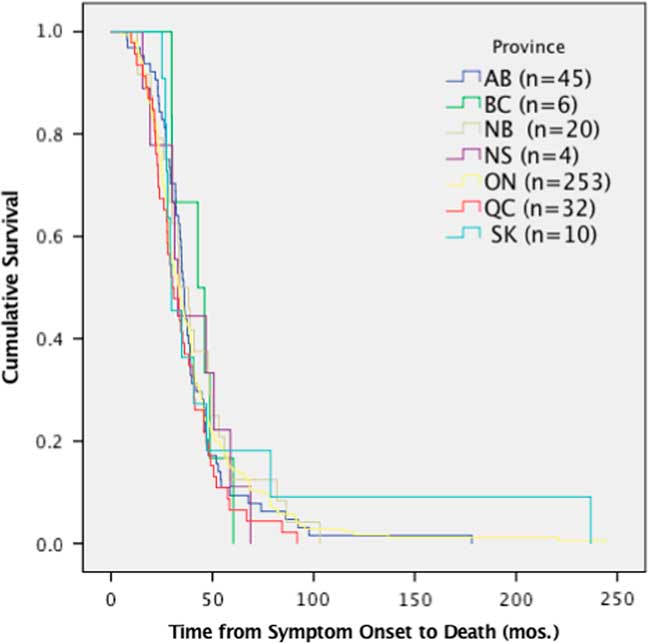
Figure 4 Survival analysis comparing median time of survival by province. Time from symptom onset to death was not significantly different per province, p>0.05 (p=0.167, χ 2=9.113). AB=Alberta; BC=British Columbia; mos=months; NB=New Brunswick; NS=Nova Scotia; ON=Ontario; QC=Quebec; SK=Saskatchewan.
Interpretation
We report the first nationwide Canadian data on time from symptom onset to diagnosis and treatment with riluzole in patients with ALS. Along with a recent paper,Reference Jackson-Tarlton, Benstead and Doucette 39 we also report nationwide Canadian data on feeding tube use. Time to diagnosis was significantly different among provinces, with Saskatchewan having the longest (27 months) and Nova Scotia having the shortest (15.1 months). Interestingly, population-based registries report time from symptom onset to diagnosis from 10 to 14 months,Reference Takei, Tsuda, Takahashi, Hirai and Palumbo 37 , Reference Kollewe, Mauss, Krampfl, Petri and Dengler 40 - Reference Chio, Mora, Calvo, Mazzini, Bottacchi and Mutani 42 whereas our consent-based registry reports a mean time to diagnosis of 21 months. It is possible that this discrepancy may partially be explained by patient recruitment to the registry in speciality ALS hospital-based clinics, resulting in slight underrepresentation of faster-progressing patients, who are in turn diagnosed more rapidly. This is supported by mean ALSFRS-R progression rates in our cohort slightly slower than the reported mean.Reference Takei, Tsuda, Takahashi, Hirai and Palumbo 37 , Reference Chio 38
Although there are some promising therapies for ALS on the horizon, the rapid disease progression and delayed time to diagnosis often result in patients being ineligible for clinical trials.Reference Zoccolella, Beghi and Palagano 10 , Reference Swash 11 , Reference Househam and Swash 14 An increasing number of studies in animal models have demonstrated that those earlier in the course of ALS symptom progression respond better to treatment.Reference Gurney, Fleck, Himes and Hall 43 - Reference Kennel, Revah and Bohme 46 Similarly, post-hoc analyses of riluzole trials have demonstrated increased efficacy in those with milder symptomology.Reference Riviere, Meininger, Zeisser and Munsat 47 Time to diagnosis is affected by several components of the ALS journey including barriers to obtaining a primary care assessment, lack of knowledge of ALS symptomatology by primary care providers and other specialists and delayed referral to a neurologist. Further delays often occur when patients are re-referred to a neuromuscular subspecialist from a general neurologist or directly to a multidisciplinary ALS clinic for confirmatory evaluation.
Furthermore, it is known that geographic remoteness can affect access to care.Reference Alari, Lafortune and Srivastava 48 - Reference Chondur, Qin, Guthridge and Lawton 50 Existing Canadian and American studies evaluating inequality in health care based on geographic variation identified factors contributing to such variation including patient need, patient preferences, illness burden, insurance coverage and community wealth/poverty.Reference De Oliveira, Ptaky and Bremner 51 - Reference Campbell, Manns, Soril and Clement 55 Further research is required into possible medical practice and health system barriers, such as referral wait times, misdiagnoses, geographic distance to clinic and urbanity that may result in longer times to diagnosis.
Although riluzole use averaged across provinces (68%) was consistent with recent studies in other countries (60%),Reference Traynor, Alexander, Corr, Frost and Hardiman 3 it was found to be statistically different among provinces with British Columbia, Saskatchewan and Alberta, all below the Canadian average (Figure 2). This may relate to several factors including the treating physician’s opinion on the benefit of riluzole, patient perceptions on potential adverse side effects or barriers arising from insurance coverage, which is province-dependent (Figure 5). Inconsistent riluzole coverage by province may only partially explain the observed differences in its usage; for example, riluzole use was the lowest in British Columbia (18%) despite its coverage through the provincial formulary and the Fair Pharmacare Program. There may be multiple possible contributors to this discrepancy: deductible payments may be unaffordable, alternative pharmaceutical or non-pharmaceutical symptom management, a more widely held patient perspective on not prolonging a diminishing quality of life or the prescribing physician’s perspectives on the benefits of riluzole. Interestingly, a recent study evaluating cost-sharing models across provinces of Canada found variation in out-of-pocket expenses for medications owing to province of residence, along with income and age.Reference Campbell, Manns, Soril and Clement 55 Saskatchewan’s limited drug coverage for riluzole during the time of analysis may have contributed to its below-average usage (52%); however, as the sample size in this province is limited, caution in interpretation is warranted. Further investigations into the motivations and barriers to riluzole use by province are required.

Figure 5 Riluzole coverage by province. Provincial formulary criteria are color-coded and displayed on the map of Canada. Saskatchewan (SK) added Riluzole to the provincial formulary in June 2017, during the period of data collection. AB=Alberta; BC=British Columbia; FVC=forced vital capacity; MB=Manitoba; NB=New Brunswick; NS=Nova Scotia; ON=Ontario; QC=Quebec; yrs=years.
Similarly, feeding tube use among provinces was statistically significant, with the lowest percent usage in British Columbia (16%) and highest in Nova Scotia (53%). A recent study evaluated factors correlated with feeding tube use in 635 ALS patients in the Canadian population,Reference Jackson-Tarlton, Benstead and Doucette 39 and found associations with dysphagia and respiratory status. Although there were no differences in overall ALSFRS-R progression rates or in the percentage of patients with bulbar onset or ventilation use by province in our study (data not shown), variation in patient characteristics may contribute to these findings. It will be important to evaluate factors affecting differences in feeding tube use in Canada in our patient population. In addition, given the variability in the referral process for feeding tubes across Canada,56 it will be essential to review guidelines and recommendations for feeding tube insertion to standardize care and outcomes across Canada.
The data in this study demonstrate that neither varying times to diagnosis nor differences in the use of riluzole or feeding tubes results in significant differences in survival rates among provinces. The patients reported here are all seen at multidisciplinary clinics, which is known to confer some survival advantage for ALS patients.Reference Rooney, Byrne and Heverin 2 , Reference Traynor, Alexander, Corr, Frost and Hardiman 3 This may account for the lack of significant difference in survival among provinces. Similarly, as data included patients newly diagnosed through 2017, it is expected that median survival will change over time, as the incident cases progress through to death. As the CNDR continues to register participants and the Canadian ALS Research Network publishes the Canadian ALS best practice guidelines (which are currently under development), time to diagnosis, riluzole, feeding tube use and survival by province will be important metrics to monitor.
These results should be interpreted in consideration of the limitations of the methodology used. There is the possibility of selection bias in the process of obtaining informed consent for recruitment into the CNDR as some participants may not provide consent excluding them from the data. In addition, selection bias may occur as some individuals are not followed up by ALS clinics. One aim of the CNDR is to improve recruitment of ALS patients and prospectively re-evaluate.
Conclusion
In addition to the recent study of feeding tube use and nutritional recommendationsReference Jackson-Tarlton, Benstead and Doucette 39 in Canada, this study contributes another “first look” at the Canadian ALS population. It demonstrates the need for further investigation of barriers to riluzole use and time to diagnosis across Canada to equalize ALS patient access to more timely care and improve clinical outcomes in this terminal disease. Similarly, studies investigating standard interventions including ventilation use and access to experimental therapies across provinces are needed.
Acknowledgments
Amyotrophic lateral sclerosis data collection by the CNDR is funded by a grant from ALS Canada (Toronto, ON). The CNDR was founded through a contribution from the Marigold Foundation (Calgary, AB).
Statement of Authorship
VLH and LK performed the statistical analyses. VLH, JL, AM and LK wrote the manuscript. TB, KS, SW, CO’C, WH, GL, MM, HB, AG, IG, WSJ, SK, CS, ST, RM, LZ and LK recruited patients and collected data. TB, KS, SW, CO’C, AM, WH, GL, MM, HB, AG, IG, WSJ, SK, CS, ST, LZ, VH, JL and LK reviewed the manuscript and provided feedback.
Disclosure
VLH, JL, AM, AG, HB, IG, WH, WSJ, SK, GL, RM, MM, CS, ST and LZ have nothing to disclose. TB reports other from Cytokinetics, outside the submitted work. CO’C reports other from Cytokinetics, outside the submitted work. KS reports grants and personal fees from Genzyme, personal fees from EMD Serono and grants from Allergan, outside the submitted work. SW reports other from Cytokinetics, outside the submitted work. LK reports grants from ALS Canada and grants from Marigold Foundation, during the conduct of the study; grants and personal fees from Biogen, grants and personal fees from Genzyme, grants from Jesse’s Journey, grants from Muscular Dystrophy Canada, grants from CIHR, personal fees from Pfizer and personal fees from Sarepta, outside the submitted work.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2018.311










 Open access
Open access