


Astroviruses (AstVs), family Astroviridae, are enteric viruses associated with gastroenteritis in young children in both developed and developing countries [Reference Guix1]. AstVs have a single-stranded positive-sense RNA genome containing three open reading frames (ORFs). ORF1a and ORF1b, at the 5′-end of the genome, encode the non-structural viral proteins (serine protease and RNA-dependent polymerase), while ORF2, at the 3′-end, encodes the capsid precursor protein. Human AstVs (HAstVs) are genetically and antigenically heterogeneous and include a major group, classified into eight antigenic types (HAstV-1 to HAstV-8) [Reference Koopmans2] and a number of unusual AstVs (e.g. MLB1, HMO, VA) discovered recently and genetically more related to animal AstVs than to ‘classical’ HAstVs [Reference Bányai3]. Sequence analysis of short fragments at either the 5′- or 3′-end of ORF2 and RT–PCR genotyping protocols with type-specific primers, have been used for genetic characterization of HAstV types 1–8 [Reference Mustafa, Palombo and Bishop4] and found to correlate with antigenic characterization with type-specific polyclonal or monoclonal antibodies [Reference Noel5]. Genotyping surveys have shown that HAstV-1 is the most common type identified in children, followed by HAstV-2, -3, -4 and -5 [Reference Gabbay6]. Interestingly, upon molecular analysis of the ORF2, discrete sequence variation has been observed within each HAstV type. HAstV-1 has been classified into four lineages (1a–1d), while HAstV-2 and HAstV-4 have been classified into three (2a–2c) and two lineages (4a, 4b), respectively [Reference Gabbay6, Reference Colomba7]. Predominant types and/or lineages appear to vary seasonally and geographically but only a few structured, nationwide epidemiological studies can be found in the literature. In addition, the use of different diagnostic and typing tools has generated heterogeneous data that are of difficult interpretation/comparison. Surveillance studies conducted in Palermo, Italy, in 1999–2000 and 2002–2005, revealed that yearly prevalence of HAstVs in children hospitalized for gastroenteritis ranged from 0·5% to 7%, similarly to the prevalence observed in other countries [Reference Colomba7–Reference De Grazia9]. The predominant type circulating in the local population was HAstV-1 with temporal fluctuations of lineages 1b and 1d. In 2002, the onset of new HAstV strains of types 2c and 4b was observed.
In the present study, we extended the investigation on the prevalence and genetic diversity of HAstVs in Italy by enrolling laboratories located in three different geographical areas. From January 2008 to December 2009 a total of 1321 faecal samples, 693 in 2008 and 628 in 2009, were obtained from children aged <5 years, hospitalized with acute gastroenteritis. Of the 2008 samples, 258 were collected in Parma (northern Italy), 283 in Bari (southern Italy), and 152 in Palermo (Sicily), while in 2009 the samples from the three areas were 216, 103 and 309, respectively. The samples were screened for the presence of HAstVs by RT–PCR with HAstV-specific primers Mon269 and Mon270, amplifying a 348 nucleotide (nt) portion at the 5′-end of ORF2 [Reference Mustafa, Palombo and Bishop4]. The amplicons were sequenced and phylogenetical analysis was performed with CLUSTALW and MEGA 5.0 (www.megasoftware.net) software using a selection of reference sequences. The sequences of Italian HAstV strains have been deposited in GenBank under accession numbers: JQ434370–JQ434393 and JQ434395–JQ434397 (for Bari); JQ434362–JQ434369 and JQ434394 (for Palermo); JQ303020, JQ303022–JQ303026 and JQ303028–JQ303034 (for Parma).
The results of our screening showed an overall rate of 3·70% (49/1321) of HAstV infections, with a higher (5·8–7·4%) range in Bari. In particular, during the 2-year study period, HAstVs were detected in Parma in 2·7% of the samples, in Bari in 6·9% and in Palermo in 1·9% of patients. A peak in HAstV prevalence was observed in Bari in 2008. Yearly prevalence, genotypes and lineages of the Italian HAstVs in the three study areas are reported in Table 1. The prevalence rates were similar to those reported in previous European studies [Reference Guix1, Reference Jakab10]. Age-stratified serological investigations have revealed seroprevalence rates ranging form 10% to 91% in children, with exposure being highest to HAstV-1 and increasing with age [Reference Koopmans2]. Accordingly, it is likely that the majority of HAstV infections do not require hospitalization or medical care and may remain undetected. A phylogenetic tree based on the 348-bp fragment of the ORF2 was constructed to investigate the genetical relationships among the HAstVs detected in this study and to compare with other published sequences. Four different genotypes (HAstV-1, HAstV-2, HAstV-4, HAstV-5) were found to have been circulating in Italy during 2008–2009. HAstV-1 was predominant, accounting for 85·7% of the HAstV strains detected in the study, followed by HAstV-2 (8·16%), HAstV-4 (6·1%) and HAstV-5 (2·04%). Phylogenetical analysis allowed us to identify clearly the various lineages and genotypes (Fig. 1). In 2008, 58·3% of HAstVs belonged to the HAstV-1d lineage, 27·8% to HAstV-1a, 8·3% to HAstV-4a and 2·7% to HAstV-2c and HAstV-5, while in 2009 HAstV-1a was predominant (80%), and apparently replaced the HAstV-1d lineage. Previous surveillance studies conducted in Palermo during 10 years have demonstrated the emergence and/or re-emergence of strains belonging to different HAstV lineages over time. In particular, HAstV-1d was first detected in Palermo in 1999, and was replaced by HAstV-1b in 2000. This lineage re-emerged in 2003–2005 and continued to be detected in 2008 along with HAstV-1a, which became predominant in 2009. Data are not available on the HAstV types circulating in other Italian areas in the same period and therefore this pattern could reflect either local or sampling variations. Furthermore, as the strains analysed in this study were collected from children with severe disease requiring hospitalization, they do not reflect the actual distribution of HAstV strains in the child population. However, our data are in agreement with other studies demonstrating that HAstV-1 is the prevalent type worldwide [Reference Koopmans2].

Fig. 1. Phylogenetical analysis of partial nucleotide sequences (348 bp) of HAstV ORF2 capsid region of 49 strains collected in Bari, Palermo, and Parma from 2008 to 2009. The Kimura two-parameter model of substitution and the neighbour-joining method were used to construct the phylogenetic tree. Bootstrap values above 70%, estimated with 1000 pseudoreplicate datasets, are indicated at each node. Italian strains of this study are indicated as follows: ▪, HAstV from Bari; ▴, HAstV from Palermo; •, HAstV from Parma.
Table 1. Temporal and geographical distribution of HAstV genotype lineages
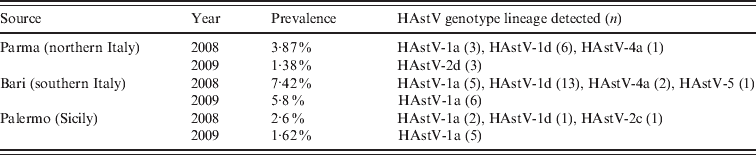
During this survey, an additional three other genotypes (HAstV-2, -4, -5) were also detected but at low frequency. HAstV-4 and HAstV-5 have been sporadically detected in different geographical areas, and are regarded as strains of minor epidemiological relevance [Reference Guix1, Reference Mustafa, Palombo and Bishop4]. On the other hand, HAstV-2 represented the second most prevalent type detected in this study and in 2009 it appeared to be more common than HAstV-1 in the area or Parma. These findings mirror the results of previous studies performed in Chile, Spain, Colombia and Brazil, describing the predominance of non-type 1 HAstVs in temporally and geographically restricted studies [Reference Victoria11]. Upon sequence analysis, two different HAstV-2 lineages were identified. The type 2c lineage was first identified in Palermo in 2002 [Reference Colomba7] and found to be circulating in 2008 in the local population, although at low frequency. The HAstV-2c strains circulating in 2008 showed 100% nucleotide identity to the old type 2c strains (PA65/02-like) (Fig. 1). However, only a small (348 bp) conserved portion at the 5′-end of ORF2 was analysed and used to predict HAstV types and lineages. Extending the analysis to the full-length ORF2 gene, including the hypervariable region of the capsid protein, could help assessing more properly the extent of genetic variation in these strains. Three type 2 strains detected in Parma in 2009 clustered together in a separate branch. Following previously defined criteria for lineage designation/distinction [Reference Guix1, Reference Noel5] we propose that these strains are designated as a new lineage, namely HAstV-2d. These novel HAstV-2d strains shared 96·7–98·8% nucleotide identity to each other and displayed less than 93% nucleotide identity to HAstV-2a, -2b and -2c strains. By observing in detail the nucleotide variation and fitting these changes into a polymorphism model for HAstV-2 lineages [Reference De Grazia8], a single nucleotide position (B381 → A) was able to differentiate the new HAstV-2d lineage from the other HAstV-2 lineages. An additional five silent nucleotide substitutions (C279 → T, C471 → T, R477 → C, Y480 → A, A549 → T), not included in the classification scheme, were helpful in distinguishing HAstV-2d from the other type 2 lineages in the short capsid region.
In conclusion, we established a nationwide surveillance system for HAstV in Italy including three different laboratories and adopted a common standardized diagnostic protocol demonstrating the co-circulation of several HAstV types and/or lineages. HAstV-1 was found to be predominant throughout the study period. A novel lineage, proposed as HAstV-2d, was found to have emerged in Parma in 2009. Investigating systematically the genetic variability of HAstVs will be important for understanding if periodic genotype shifts and genetic/antigenic drifts occur and if these changes may fit a model of evolution analogous to those observed for other antigenically/genetically heterogeneous viruses.
ACKNOWLEDGEMENTS
This investigation was supported by the grant ‘Ricerca Scientifica FIL 2009’, from the University of Parma, Italy.
DECLARATION OF INTEREST
None.


