


The discovery that a significant proportion of cellular proteins do not have a well-defined structure under physiological conditions is one of the most fascinating concepts in modern biology (Ref. Reference Wright and Dyson1). This iconoclastic finding has shaken the broadly accepted dogma that a protein is predisposed to fold into a unique structure that is specified by its amino acid sequence.
Proteins that lack a well-defined 3D structure have been referred to as ‘intrinsically disordered proteins’ (IDPs), and they constitute the part of the proteome called the ‘unfoldome’ (Ref. Reference Uversky2). IDPs perform a wide range of regulatory functions related to signal transduction, molecular recognition and molecular assembly (Ref. Reference Dunker and Uversky3), and several have been implicated in fatal human pathologies (Ref. Reference Uversky4). One representative member of IDPs is α-synuclein (encoded by SNCA), a brain protein associated with Parkinson disease. Depending on its environment, α-synuclein can adopt a variety of structurally unrelated conformations (Fig. 1), from a natively unfolded (mostly disordered) state to different α-helical or β-structural species folded to a different degree (Ref. Reference Uversky5). The protein can also self-aggregate and form various types of supramolecular assemblies with different morphology, including oligomers, amorphous aggregates and amyloid-like fibrils (Ref. Reference Uversky5). This incredible level of conformational plasticity is also a hallmark of other amyloidogenic proteins, such as amyloid β (Aβ; encoded by APP) involved in Alzheimer disease and the prion protein (PrP; encoded by PRNP) involved in Creutzfeldt–Jakob disease and prion infections (Ref. Reference Ghahghaei and Faridi6). Some amyloidogenic proteins (Alzheimer Aβ peptides, α-synuclein) belong to the IDP family; in other cases (e.g. PrP), the protein has a 3D structure but can misfold into an alternative structure that is prone to aggregation. There are several mechanisms by which an amyloidogenic protein can acquire a disease-related 3D structure: overexpression, mutations or other genetic alterations, and exposure to harmful environmental conditions (Ref. Reference Uversky2). These mechanisms have been extensively studied during the past two decades. However, how amyloidogenic proteins induce cell death in these neurodegenerative diseases is still a mystery. Given the high prevalence and the fatal outcome of these pathologies in humans, this is clearly one of the most important challenges of biomedical research for the 21st century.

Figure 1. Different pathways of amyloidogenic protein oligomerisation and fibrillation on membrane surfaces. Upon interaction with neuronal membranes, unstructured soluble monomers of amyloidogenic proteins undergo an α-helix shift of their conformation (a). Further accumulation of the proteins on the surface of the membrane induces oligomerisation into β-sheet aggregates (b) or α-oligomers (c). Oligomers with a β-sheet structure can form protofibrils (d), amyloid fibrils (e) and amyloid pores (annular protofibrils) with ion-channel properties (f). In some instances (e.g. for α-synuclein), transmembrane channels can also be generated by oligomers with an α-helix structure (g). In all cases, the formation of functional ion channels requires the insertion and assembly of the oligomers in the neuronal membrane (green lipid bilayer). Note the possibility of conversion between α- and β-oligomers (h).
Among the different research strategies in this field, those aiming at identifying common features in distinct amyloidogenic proteins have been particularly fruitful. Obviously, it is somewhat paradoxical that the similar behaviour of amyloidogenic proteins – that is, their conformational plasticity and aggregation properties – does not rely on any kind of homology in their amino acid sequence. Instead, these common features might involve the 3D structures of these proteins. A major breakthrough was the identification of a common epitope in oligomers formed by various amyloidogenic proteins (Ref. Reference Kayed7). This discovery suggested a universal mode of oligomerisation underlying a common mechanism of toxicity. Moreover, conformation-dependent antibodies that recognise generic epitopes have been used as a structural basis to distinguish amyloid prefibrillar and fibrillar oligomers (Ref. Reference Glabe8). A second breakthrough was the demonstration that amyloidogenic proteins can assemble into annular structures forming amyloid pores with ion channel properties (Refs Reference Lashuel9, Reference Quist10) (Fig. 1). This finding suggested that amyloid oligomers could perturb membrane permeability (e.g. cellular calcium fluxes) and thus kill neuronal cells more efficiently than amyloid fibrils. This mechanism, originally proposed by Arispe et al. (Refs Reference Arispe, Rojas and Pollard11, Reference Arispe, Rojas and Pollard12) for explaining the toxicity of Aβ, is now commonly referred to as the β-amyloid calcium channel hypothesis (Ref. Reference Vestergaard, Hamada and Takagi13). However, despite their fruitful contribution to the field, these important discoveries did not really inform us on the molecular mechanisms involved in the different modes of amyloid oligomerisation and aggregation. To progress towards this knowledge, we have to consider another common feature tightly linked to the biology of amyloids in the brain, which lies in their immediate environment – that is, biological membranes (Refs Reference Vestergaard, Hamada and Takagi13, Reference Munishkina and Fink14). Indeed, Aβ peptides are generated by the proteolytic cleavage of a transmembrane precursor (the amyloid protein precursor, APP) (Ref. Reference Hardy and Higgins15), PrP is a membrane protein anchored in the external leaflet of the plasma membrane by a glycosylphosphatidylinositol anchor (Ref. Reference Prusiner16), and α-synuclein is involved in the traffic of synaptic vesicles (Ref. Reference Cabin17).
Amyloidogenic proteins reside in a lipid world constituted by a myriad of lipid-based structures such as internal membrane networks, plasma membrane nano- and microdomains, lipoproteins and exosomes. This is not totally surprising if one considers that lipids are the most abundant organic compounds found in the brain, accounting for up to 50% of its dry weight (Ref. Reference Fantini and Barrantes18). As in other mammalian tissues, brain lipids consist of three major categories in roughly equimolar proportions (Ref. Reference Fantini19): glycerophospholipids, sphingolipids and cholesterol. Sphingolipids and cholesterol self-aggregate into specific membrane domains referred to as nanodomains, microdomains, caveolae, lipid shells or lipid rafts (Refs Reference Simons and Ikonen20, Reference Lingwood and Simons21). These sphingolipid–cholesterol-enriched domains (the so-called lipid rafts) are in a liquid-ordered (Lo) phase floating in the more liquid glycerophospholipid-rich and cholesterol-poor bulk (Ld phase) of the plasma membrane. Lipid rafts are chiefly involved in cellular trafficking and signalling functions (Ref. Reference Drevot22) and are also preferential sites for host interactions with pathogens and toxins (Ref. Reference Taïeb, Yahi and Fantini23).
The aim of this review is to emphasise the specific role of cholesterol and sphingolipids, alone and in combination, in the interaction between amyloidogenic proteins and neural membranes, and to figure out how these interactions can affect the structural and functional properties of these proteins. We also discuss the impact of age- and disease-related changes in brain lipid content on the pathophysiology of amyloid-associated neurodegenerative diseases. We do not intend to present a separate section for each amyloidogenic protein (Aβ, α-synuclein, PrP, etc.). Instead, it is our goal to identify common membrane-based mechanisms that, at least at the molecular level, might apply to any type of amyloidogenic protein and thus could serve as a solid biochemical background for studying amyloid toxicity in the brain.
Mechanisms of concentration of amyloidogenic proteins on neuronal membranes
Reduction of dimensionality
There are two main mechanisms ensuring the efficient transfer of amyloidogenic proteins from the cytosolic or extracellular milieu to neuronal plasma membranes. The first one is the reduction of dimensionality (from 3D to 2D) that occurs when a soluble protein dissolved in the bulk solvent binds to a membrane surface (Ref. Reference Adam, Delbrück, Rich and Davidson24). The underlying idea is that the membrane 2D space can concentrate proteins far more efficiently than the 3D space of the bulk solvent. As a consequence, the average distance between two proteins is shorter in 2D than in 3D, and this effect is more pronounced for higher protein concentrations (Ref. Reference Aisenbrey25). This means that increased concentration of a protein lowers the average protein–protein distance more efficiently on a 2D surface than in a 3D space. Consequently, protein–protein interactions are favoured in the 2D space. This phenomenon is particularly important for amyloidogenic proteins that tend to self-aggregate after a conformational transition (Fig. 2). Thus, the simple fact of the concentration of monomers in a restricted membrane area is, by itself, a crucial step in the complex process of amyloid oligomerisation/aggregation.
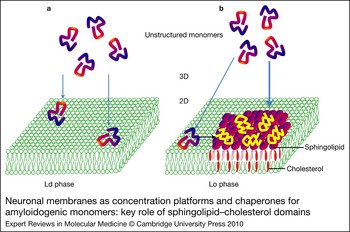
Figure 2. Neuronal membranes as concentration platforms and chaperones for amyloidogenic monomers: key role of sphingolipid–cholesterol domains. (a) Amyloidogenic monomers have a low affinity for the liquid-disordered (Ld) phase of plasma membranes enriched in phosphatidylcholine. (b) By contrast, the monomers have a high affinity for sphingolipid–cholesterol domains in the liquid-ordered (Lo) phase. In this case, monomers are not only concentrated on the membrane surface, but also undergo a major conformational change induced by the sphingolipids. This property is referred to as the chaperone effect of sphingolipids on amyloidogenic proteins.
Specific binding to membrane lipids
The second mechanism explaining the affinity of amyloidogenic proteins for neuronal membranes is lipid specificity. Converging data obtained from various laboratories with a broad range of experimental approaches have shown that amyloidogenic proteins interact with the lipids found in the Lo phase – that is, sphingolipids and cholesterol (Fig. 2). Alzheimer Aβ peptides recognise several glycosphingolipids, including neutral species such as asialo-GM1 or galactosylceramide (GalCer) as well as gangliosides such as GM1 (Refs Reference Valdes-Gonzalez, Inagawa and Ido26, Reference Mahfoud27, Reference Hebbar28, Reference Yanagisawa29); α-synuclein binds to gangliosides GM1 and GM3 (Refs Reference Martinez30, Reference Di Pasquale31); PrP interacts with sphingomyelin, GalCer, GM1 and GM3 (Refs Reference Mahfoud27, Reference Mattei32, Reference Sanghera and Pinheiro33) and is associated with sphingolipid signalling platforms (Ref. Reference Schmalzbauer34). Overall, this high affinity for sphingolipids determines the concentration of amyloid proteins in lipid raft areas of the extracellular leaflet of the plasma membrane. Several amyloidogenic proteins can also bind to negatively charged glycerophospholipids (Refs Reference Jha35, Reference Jo36), which are enriched in the cytoplasmic leaflet of the plasma membrane (Ref. Reference Fantini19). This type of interaction has been extensively studied for α-synuclein, which recognises anionic phosphatidylserine and phosphatidylglycerol (Refs Reference Jo36, Reference Ramakrishnan, Jensen and Marsh37, Reference Kubo38, Reference Stöckl39). In this review, we restrict our analysis to the role of cholesterol and sphingolipids in the interaction of amyloidogenic proteins and membranes. As we will see, these lipids play a more active role in the conformation, oligomerisation and aggregation of amyloidogenic proteins than just acting as a concentration platform.
Molecular mechanisms of sphingolipid–amyloidogenic-protein interactions
Structural diversity of membrane sphingolipids
That the same amyloidogenic protein could interact with several distinct sphingolipids (Refs Reference Valdes-Gonzalez, Inagawa and Ido26, Reference Mahfoud27, Reference Di Pasquale31) could be viewed as paradoxical and difficult to reconcile with a real specificity of interaction. A biochemical analysis of sphingolipid structures is necessary to get a clearer view on this issue. Sphingolipids are formed by the condensation of a fatty acid chain with a sphingoid base (which is generally sphingosine although five different sphingoid bases have been detected in mammalian cells). The biochemical diversity of sphingolipids is first generated by the numerous fatty acids (more than 20, varying in chain length, degree of saturation and degree of hydroxylation) that can be attached to the sphingoid base to form a ceramide (Ref. Reference Futerman and Hannun40). Sphingolipids are then classified into sphingomyelin and glycosphingolipids, according to the biochemical nature of the polar part attached to ceramide: phosphorylcholine for sphingomyelin, and a glycan moiety for glycosphingolipids. Several hundred different carbohydrate structures have been described in glycosphingolipids, with various combinations of neutral sugars, sulfated sugars and sialic acids (Ref. Reference Fantini19).
Cracking the code governing glycosphingolipid–protein interactions
Despite this huge structural diversity, glycosphingolipids can share common structural fingerprints that render them ‘readable’ by the same protein. This is the case for the glycosphingolipids GalCer, globotriaosylceramide (Gb3) and GM3, which have a distinct sugar moiety but a common galactosyl residue with the same orientation (Fig. 3a–c). As a logical consequence of this common structural feature, a protein that recognises one of these glycosphingolipids (e.g. GalCer), through binding to this galactose residue, might also interact with the other two (in this case, Gb3 and GM3). This means that the biochemical code governing glycosphingolipid–protein interactions is degenerate. An example of this striking property is the V3 loop domain of the human immunodeficiency virus 1 (HIV-1) surface-envelope glycoprotein gp120, which was shown to recognise first GalCer (Refs Reference Harouse41, Reference Yahi42, Reference Cook43), and then, several years later, both Gb3 and GM3 (Refs Reference Hammache44, Reference Hammache45). However, there is a unique molecular mechanism that accounts for the interaction of each of these glycosphingolipids with gp120 (Ref. Reference Delézay46), and this mechanism also applies for amyloidogenic proteins.

Figure 3. Glycosphingolipids and amyloidogenic proteins: a common mechanism of interaction? Despite their high level of biochemical diversity, some glycosphingolipids share a common galactosyl ring with an orientation compatible with the establishment of stacking CH-π interactions between the sugar and an aromatic residue. Such galactosyl rings are indicated in shaded circles in the sugar moiety of ganglioside GM3 (a), globotriaosylceramide Gb3 (b) and galactosylceramide GalCer (c). In these cases, the apolar side of the galactosyl ring fits particularly well with the aromatic-ring side chain of an aromatic residue (Phe, in this case) exposed at the surface of an amyloid protein (d). The electronic cloud of the π system stacks onto the CH groups of the galactosyl ring. This interaction is favoured by the common geometric structure of both rings, which allows a coordinated binding process between CH groups and π electrons. Cholesterol, which has a strong affinity for sphingolipids, can further improve the accessibility of the galactosyl group through fine tuning of glycosphingolipid conformation (e). This effect involves the establishment of an H-bond network between the OH group of cholesterol, the NH group of the sphingolipid and the oxygen atom of the glycosidic bond linking the ceramide and the sugar part of the sphingolipid. GalCer-NFA, GalCer with a nonhydroxylated fatty acid in the ceramide moiety.
Let us first consider the way in which proteins bind to GalCer, which is both the major lipid of myelin sheaths (Ref. Reference Curatolo47) and the simplest possible brain-expressed glycosphingolipid, consisting of a ceramide linked to a single galactosyl group (i.e. a ceramide monohexoside). Most of the GalCer molecule is dipped into the membrane, so that the galactosyl moiety is the principal region of the glycolipid available for protein binding. In this respect, there is a functional similarity between proteins that recognise GalCer and galactose-specific lectins. However, the local concentration of galactosyl groups in a nanodomain of GalCer is extremely high compared with the maximal concentration of free galactose in biological fluids (another illustration of the reduction-in-dimensionality concept discussed above). This explains why even high concentrations of free galactose cannot inhibit GalCer–gp120 interactions (Ref. Reference Harouse41). Moreover, the conformation of the galactose head group in the glycolipid is restricted by the ceramide backbone, which allows only limited motion of the sugar (Ref. Reference Nyholm, Pascher and Sundell48). By contrast, galactose in water solution is free to adopt a wide range of conformations. Finally, the most polar part of ceramide, which includes both amide and hydroxyl (OH) groups, as well as the β-glycosidic bond linking ceramide to galactose, can also participate in the binding reaction and further stabilise the GalCer–protein complex. Taken together, these biochemical properties of the galactosyl head group of GalCer explain why GalCer–protein interactions are distinct from galactose–lectin interactions (Ref. Reference Fantini49).
Key role of aromatic residues in protein–glycosphingolipid interactions
At the molecular level, numerous proteins that bind to GalCer have a solvent-exposed aromatic residue that provides a complementary surface for a stacking interaction with the galactose ring (Fig. 3d). As a result of the distribution of the OH groups linked to the pyranosyl ring of galactose, this sugar has two chemically distinct faces: one apolar with hydrocarbyl (CH) groups and the other polar with OH groups. Therefore, the apolar face of galactose is particularly suited for a stacking interaction with an aromatic structure (Ref. Reference Maresca50). This type of interaction between CH groups and a π-electron cloud system has been described and referred to as CH–π interaction by Nishio and co-workers (Ref. Reference Nishio51). The CH–π stacking mechanism is involved in the interaction between glycosphingolipids and several amyloidogenic proteins, including PrP (Ref. Reference Fantini49), Aβ (Ref. Reference Yahi, Aulas and Fantini52) and amylin (Ref. Reference Levy53). In both Gb3 and GM3, the apolar side of the central galactosyl group has a similar orientation to that in GalCer (Fig. 3a–c); thus, GalCer-binding proteins are predicted to also bind to Gb3 and GM3 through CH–π stacking. Note that the conformation of the sugar moiety of glycosphingolipids is restricted by a network of H-bonds (Ref. Reference Yahi, Aulas and Fantini52) that involves either an OH group in the acyl chain of the ceramide (intramolecular network) or the OH group of cholesterol (intermolecular network, Fig. 3e), as further discussed below (see the section ‘The outside and inside effects of cholesterol’).
The notion that unrelated proteins, displaying a solvent-accessible aromatic residue in a hairpin domain, might bind these glycosphingolipids through a similar stacking mechanism has motivated the search for V3-like domains in various proteins, including amyloidogenic proteins (Ref. Reference Mahfoud27). This is the concept of the glycosphingolipid-binding domain or, more generally, of the sphingolipid-binding domain (Ref. Reference Fantini49). Such domains are characterised by the presence, in a flexible region of the surface of a protein, of an aromatic residue with its side chain oriented towards the solvent, as illustrated in Figure 4a with the superimposition of the glycosphingolipid-binding domains of Aβ and α-synuclein (Ref. Reference Di Pasquale31). The recognition of the glycosphingolipid by this region of the protein is thought to proceed through an induced-fit mechanism involving the conformational mobility of the aromatic side chain and a concomitant adjustment of the orientation of the galactosyl head group (Fig. 4b).

Figure 4. A common sphingolipid-binding domain in amyloidogenic proteins. (a) Amyloidogenic proteins share a common sphingolipid-binding domain, which can be shown by superimposing the structure of micelle-bound Alzheimer Aβ peptide [PDB ID: 1BA4, in red] (Ref. Reference Coles150) onto the structure of micelle-bound α-synuclein [PDB ID: 1XQ8, in blue] (Ref. Reference Ulmer142). The motif corresponds to a helix–turn–helix structure displaying an aromatic residue (Tyr10 for Aβ; Tyr39 for α-synuclein) oriented towards the solvent and located at a similar position in the loop. The location of Glu residues associated with Glu to Lys mutations in genetic forms of Alzheimer and Parkinson diseases is indicated (Glu22 for Aβ and Glu46 for α-synuclein, respectively). The amino acid sequence of both proteins (upper panel) show very little homology, apart from the above-mentioned Tyr residues (asterisk) and a common Val residue (Val52 for α-synuclein and Val24 for Aβ). (b) Due to the high conformational plasticity of amyloidogenic proteins, the sphingolipid-binding domain can adopt several distinct conformations, as shown for the core amyloid-forming motif of amylin [NFGAILSS octapeptide, PDB ID: 1KUW] (Ref. Reference Mascioni68). The α-carbon chain of the peptide is coloured in blue, and the Phe residue in red. Twenty conformers obtained by nuclear magnetic resonance (NMR) analysis are superposed. Once bound to the glycosphingolipid (GSL, coloured in green), the amyloid peptide is locked in a unique conformation. Cholesterol (Chol) is coloured in orange. This drawing is based on data in Ref. Reference Levy53 obtained with lactosylceramide (LacCer).
In summary, a glycosphingolipid-binding domain is a flexible domain containing turn-inducing (Gly, Pro), basic (Arg, Lys, His) and aromatic residues (Phe, Tyr, Trp) (Refs Reference Mahfoud27, Reference Fantini, Garmy and Yahi54, Reference Chakrabandhu55, Reference Taieb56, Reference Hamel, Fantini and Schweisguth57). The critical involvement of aromatic residues in the interaction between proteins and glycosphingolipids has been convincingly demonstrated by using synthetic peptides in which these residues were replaced by Ala or Leu (Refs Reference Levy53, Reference Fantini, Garmy and Yahi54, Reference Chakrabandhu55): correspondingly, the interaction with the glycosphingolipid has been dramatically decreased or even totally lost. Reciprocally, conservative aromatic substitutions (e.g. Phe to Trp) did not affect binding to glycosphingolipids (Ref. Reference Fantini, Garmy and Yahi54). Taken together, these data show that the sphingolipid-binding domain mediates specific protein–glycosphingolipid interactions driven by CH–π stacking of a sugar and aromatic residue (Refs Reference Yahi, Aulas and Fantini52, Reference Snook, Jones and Hannun58).
Glycosphingolipids exert a chaperone effect on amyloidogenic proteins
Conformational changes mediated by sphingolipid-binding domains
One important consequence of protein–sphingolipid interactions is that the conformation of the protein can be significantly affected on binding to sphingolipids (Fig. 4b). This chaperone effect of sphingolipids is a direct consequence of the unique modalities of protein–sphingolipid interactions (Ref. Reference Fantini49). As soon as binding begins, the structure of the sphingolipid changes and the protein gradually adapts its shape to this moving target. A stable anchoring of the protein to the sphingolipids can be obtained by optimising protein–sphingolipid contacts, and this requires significant conformational changes of the protein. The flexibility of the sphingolipid-binding domain is particularly well suited to trigger structural rearrangements in the protein (Ref. Reference Fantini49). This unexpected consequence of protein–sphingolipid interactions has been extensively studied with HIV-1. Briefly, on binding to raft glycosphingolipids through its V3 domain, HIV-1 gp120 undergoes a series of conformational changes that are required for virus fusion (Refs Reference Fantini19, Reference Hammache45). Because the sphingolipid-binding domain is evolutionarily conserved (it has been found in virus, bacteria, insect and mammalian proteins), it is likely that sphingolipids can modulate the conformation and regulate the function of a wide range of proteins (Refs Reference Yahi, Aulas and Fantini52, Reference Levy53, Reference Fantini, Garmy and Yahi54, Reference Chakrabandhu55, Reference Taieb56, Reference Hamel, Fantini and Schweisguth57).
Chaperone effect of ganglioside GM1 on Alzheimer Aβ peptides
How could this concept of a sphingolipid-mediated chaperone effect apply to membrane-bound amyloids? Seminal work by Matsuzaki, Yanagasiwa and co-workers has shown that lipid rafts act as surface catalysts able to accelerate the aggregation of Aβ (Refs Reference Yanagisawa29, Reference Kakio59, Reference Matsuzaki60, Reference Kakio61, Reference Okada62, Reference Matsuzaki, Kato and Yanagisawa63). Most interestingly, GM1-bound Aβ has been identified as a pre-amyloid form acting as an endogenous seed for the formation of neurotoxic amyloid fibrils in Alzheimer disease (Refs Reference Yanagisawa29, Reference Kakio61, Reference Okada62, Reference Matsuzaki, Kato and Yanagisawa63). These data have not only strengthened the role of lipid rafts as preferential binding sites for unstructured amyloidogenic proteins on neuronal membranes but have also suggested that these membrane domains behave as conformational catalysts for amyloid formation. In other words, lipid rafts can control the conformation of membrane-bound amyloid through a chaperone effect (Refs Reference Fantini49, Reference Kakio59, Reference Fantini64). Yet, conflicting data have suggested that binding of Aβ to GM1 induces a conformational transition from random coil to a protective α-helical structure (Ref. Reference McLaurin65), as also observed for α-synuclein (Ref. Reference Martinez30), or to a fibrillation-prone β-structure (Refs Reference Kakio61, Reference Choo-Smith and Surewicz66). The structure of amyloidogenic proteins bound to GM1 depends on several physicochemical parameters, including pH, membrane fluidity, GM1-carrier lipid ratios, protein concentration and the absence or presence of cholesterol, which could explain the discrepancies reported in the literature. In any case, the available data suggest that lipid rafts could (1) modulate the conformational changes of unstructured monomers from the unfolded state to more-ordered α or β structures, and (2) control the balance between α and β structures, which in turn determine the oligomerisation/aggregation status of amyloidogenic proteins.
Kakio et al. (Ref. Reference Kakio59) have shown that the oligomerisation and aggregation of Aβ peptides occur in ganglioside-enriched domains of the plasma membrane. A possible sequence of events could be as follows: (1) soluble Aβ monomers bind to the sugar head group of gangliosides in cholesterol–sphingolipid-enriched domains such as lipid rafts; (2) on binding to gangliosides, Aβ is constrained to adopt an α-helical structure; (3) as the protein density on the membrane increases, Aβ undergoes a conformational transition from an α-helix-rich structure to a β-strand-rich one; and (4) the ganglioside-bound Aβ complex with acquired secondary structures serves as a seed for amyloid fibril formation. Indeed, Aβ fibrils with high toxicity have been successfully generated in an acellular system using GM1-containing raft-like liposomes (Ref. Reference Okada67). Altogether, these data strongly support the concept that the ganglioside cluster can act as a chaperone able to lower the activation energy for the conformational changes of Aβ (Ref. Reference Kakio59). This scenario is also consistent with the involvement of the glycosphingolipid-binding domain in membrane–amyloid interactions (Refs Reference Levy53, Reference Fantini64).
How glycosphingolipids could stabilise an α-helix and prevent amyloid formation
Unstructured amyloidogenic proteins can adopt a wide range of conformations in water and in biological membranes (Fig. 1). The conformational flexibility of the amyloid core peptide of amylin (NFGAILSS) has been studied by nuclear magnetic resonance (NMR) (Ref. Reference Mascioni68). In a membrane-like micellar environment, this peptide can adopt three distinct classes of conformations owing to the wide flexibility of the peptide chain (Fig. 4b). Correspondingly, the unique Phe residue of the motif has three main possible orientations (Ref. Reference Levy53). On binding to a glycosphingolipid bearing an appropriately oriented galactosyl group (lactosylceramide in Fig. 4b), the Phe residue stacks onto the sugar so that only one of the numerous peptide conformations is selected and locked.
This illustrates the potency of CH–π stacking interactions to stabilise a thermodynamically unstable conformation at a minimal energetic cost, just like a wedge can efficiently block a car on a sloping street (Ref. Reference Fantini49). This simple but efficient mechanism could allow raft glycosphingolipids to act as molecular chaperones inducing α-helix-rich structures in amyloidogenic proteins (Fig. 5). In the case of PrP, the stabilisation of an α-helix structure is beneficial, because this can prevent protein misfolding and aggregation (Ref. Reference Fantini49). However, the α-helix conformation could also facilitate membrane insertion and channel/pore formation, leading to dramatic perturbations of membrane permeability, as shown for α-synuclein (Ref. Reference Zakharov69). In addition, the chaperone effect of glycosphingolipids is not irreversible. Changing the physicochemical properties of the raft–protein complex, for example through oxidative damage to the proteins or lipids (Refs Reference Ruf70, Reference Bosco71), could theoretically induce the dissociation of the amyloid peptides from glycosphingolipids such as GalCer. Then, the sudden exposure of aromatic side chains would allow a tight packing (through π–π interactions) between adjacent helices. This would lead to the formation of a dimer, a key step in the generation of amyloid fibrils (Refs Reference Knaus72, Reference Krishnan73), which is thought to proceed through an ordered polymerisation of β structures (Ref. Reference Wille74).

Figure 5. How glycosphingolipids could promote α-helix structures in amyloidogenic proteins. When bound to glycosphingolipids (GSLs) through the sphingolipid-binding domain, the aromatic residues of two vicinal amyloidogenic proteins cannot interact with each other and the protein is forced to adopt an α-helix conformation. The molecular mechanism of this interaction is the CH–π stacking between the galactosyl ring of GalCer and the aromatic residue of the protein. The α-helix structure of amyloidogenic proteins is not only present in membrane-bound monomers (as illustrated in Fig. 2, right panel), but also in oligomeric α-synuclein channels (Fig. 1g). The β-amyloid aggregation process is triggered when the glycosphingolipid–protein complex is destabilised, resulting in the release of the protein from its glycosphingolipid chaperone (a). In this new glycosphingolipid-free environment, the aromatic residues of two vicinal amyloidogenic proteins can interact together (b), inducing a drastic conformational change from an initial α-helix to a β-strand structure. Amyloid aggregation then results from the π–π-driven assembly of aromatic side chains (for a review, see Ref. Reference Gazit79), forming amyloid dimers that in turn associate to form amyloid fibrils (as illustrated in Fig. 1e).
From this discussion, we would like to underscore (1) the key role of aromatic residues in the self-assembly of amyloid fibrils and (2) the chaperone effect of raft glycosphingolipids that could induce otherwise thermodynamically unstable α-helix structures of amyloid fibril-forming proteins. Indeed, most amyloidogenic proteins (including Aβ and PrP) have an amino acid sequence that is expected to form β but not α structures (Ref. Reference Kallberg75). These proteins are referred to as ‘α-helix/β-strand discordant’. Lipid rafts can prevent those proteins from adopting the conformation they prefer, stabilising their structure in a ‘forced’ α conformation. This stabilisation can occur in the early biosynthetic pathway as demonstrated for PrP (Ref. Reference Sarnataro76). Inhibition of sphingolipid biosynthesis logically increases PrP misfolding into disease-associated β structures (Ref. Reference Naslavski77). This mechanism seems to also apply for several neurotransmitter receptors (e.g. the nicotinic acetylcholine receptor) whose transport and assembly in the plasma membrane require the assistance of sphingolipids (Ref. Reference Baier and Barrantes78). Mutations in amyloidogenic proteins that are associated with inherited forms of neurodegenerative diseases have been shown to directly affect sphingolipid binding. This is the case for the Creutzfeldt–Jakob-linked E200K mutation of PrP, which affects PrP–sphingomyelin interactions (Ref. Reference Mahfoud27).
In summary, by providing a complementary surface for amyloidogenic proteins, cellular glycosphingolipids might constitutively stabilise their α conformation and inhibit their aggregation, as recently demonstrated for amylin (Ref. Reference Levy53). Replacing these glycosphingolipid–protein CH–π interactions with protein–protein π–π interactions (Refs Reference Fantini and Barrantes18, Reference Maresca50) would then trigger amyloid growth and deposition (Refs Reference Fantini49, Reference Gazit79). To what extent glycosphingolipids could also stimulate β-strand-mediated amyloid aggregation (Ref. Reference Kakio59) remains to be described and explained at the molecular scale. The mutual effect of GM1 and Aβ on their respective conformations is likely to displace the equilibrium of the reaction towards the formation of amyloid aggregates, as observed in vitro (Ref. Reference Mao80).
Finally, it should be noted that noncovalent interactions distinct from CH–π stacking might also contribute to sphingolipid–amyloid interactions. These could include hydrophobic interactions (Ref. Reference Utsumi81), H-bonds and electrostatic bridges (especially for sulfated glycosphingolipids, gangliosides and sphingomyelin) (Ref. Reference Ikeda and Matsuzaki82). Indeed, in addition to aromatic side chains (Tyr10), several aliphatic (Ref. Reference Utsumi81) and basic residues such as His13 have been implicated in GM1–Aβ interactions (Ref. Reference Williamson83). Anionic phospholipids such as phosphatidylserine, which bind to α-synuclein through electrostatic interactions (Ref. Reference Kubo38) and stimulate the formation of amyloid fibres (Ref. Reference Zhao, Tuominen and Kinnunen84), could, in the cytoplasmic leaflet of the plasma membrane, have a role equivalent to the one played by gangliosides in the external leaflet.
The outside and inside effects of cholesterol
Cholesterol as a risk factor and a therapeutic target for neurodegenerative diseases
High cholesterol has been identified as a major risk factor for both Alzheimer (Ref. Reference Martins85) and Parkinson (Ref. Reference Hu86) diseases, and dysregulation of cholesterol homeostasis is a hallmark of neurodegenerative diseases (Ref. Reference Liu87). Unfortunately, the effects of cholesterol on amyloid fibrillogenesis and toxicity are not well understood and the results reported so far are controversial (Ref. Reference Yanagisawa88). Cholesterol directly binds to APP (Ref. Reference Beel89) and stimulates its insertion into phospholipid monolayers (Ref. Reference Lahdo and de La Fournière-Bessueille90). It also binds to Aβ protofibrils (Ref. Reference Harris91). However, whether cholesterol accelerates (Ref. Reference Harris92) or decreases (Refs Reference Arispe and Doh93, Reference Yip94) Aβ polymerisation is still uncertain. Moreover, the generation of Aβ peptides through APP proteolysis occurs within lipid rafts and is sensitive to inhibitors of cholesterol biosynthesis (Ref. Reference Hartmann95), so that the involvement of cholesterol homeostasis in Alzheimer disease cannot be simply ascribed to the regulation of Aβ fibrillogenesis.
In the case of Parkinson disease, data obtained with both cultured neuronal cells and transgenic mice have shown that the cholesterol-depleting agent methyl-β-cyclodextrin decreased the level of α-synuclein in membrane fractions (Ref. Reference Bar-On96). Moreover, metabolic inhibition of cholesterol biosynthesis with statins reduced the levels of α-synuclein accumulation in neuronal membranes, whereas cholesterol supplementation of cultured neurons increased α-synuclein aggregation (Ref. Reference Bar-On97). Consistent with these studies, it was recently reported that lovastatin treatment of α-synuclein transgenic mice was associated with a marked reduction of α-synuclein aggregation and abrogation of neuronal pathology (Ref. Reference Koob98). This suggests that treatment with cholesterol-lowering agents might be beneficial for patients with Parkinson disease. It has been suggested that oxidised cholesterol metabolites could accelerate α-synuclein aggregation (Ref. Reference Bosco71), which provides a mechanistic link between oxidative stress and Parkinson disease. Yet the molecular mechanisms by which cholesterol and/or its metabolites could affect the oligomerisation/aggregation status of amyloidogenic proteins have not been fully deciphered. Recent physicochemical data could shed some light on this complex issue.
Effects of cholesterol on sphingolipid conformation
First, cholesterol has a major impact on the conformation of sphingolipids. The apolar part of sphingolipids (i.e. the most important part of the ceramide) interacts with the smooth face of cholesterol, whereas the OH group of cholesterol is accessible to the polar part of the sphingolipid (Ref. Reference Fantini and Barrantes18). In the case of glycosphingolipids, this OH group is involved in an H-bond network that restricts the conformation of the sugar moiety in a parallel orientation with respect to the membrane. This effect is particularly important when the ceramide contains a nonhydroxylated fatty acid (NFA, Fig. 3e). Because this conformation of the sphingolipid is particularly suited for a sphingolipid-binding domain, cholesterol usually accelerates protein binding to sphingolipid (Ref. Reference Mahfoud99) and this also applies for amyloidogenic proteins (Ref. Reference Yahi, Aulas and Fantini52). By contrast, when the sphingolipid contains a hydroxylated fatty acid (HFA), the OH group of cholesterol is excluded from the H-bond network and it cannot exert its conformational effect on the sphingolipid. In this case, cholesterol does not improve but rather perturbs the organisation of sphingolipids and can even inhibit glycosphingolipid binding to amyloidogenic proteins (Ref. Reference Yahi, Aulas and Fantini52). Thus, according to the distribution of HFAs versus NFAs in sphingolipids, an increase of membrane cholesterol can lead to opposite effects on sphingolipid-mediated amyloidogenic protein binding and aggregation. In any case, these effects are due to a fine tuning of sphingolipid conformation induced by cholesterol, and do not involve any kind of physical interaction between amyloidogenic proteins and cholesterol.
Amyloidogenic proteins interact first with sphingolipids and then with cholesterol
By contrast, the second mechanism involves a direct interaction between the amyloidogenic protein and cholesterol. Therefore, this effect requires the insertion of a part of the amyloid protein in the membrane; otherwise the protein would not be in physical contact with cholesterol. Devanathan et al. (Ref. Reference Devanathan100) have shown that Aβ peptide aggregation on the bilayer surface requires a sphingomyelin-rich environment but can occur in the absence of cholesterol. This is in line with our hypothesis that sphingolipids are fully responsible for the initial binding of amyloidogenic proteins to lipid rafts, and that cholesterol is not directly involved at this stage. However, at the second step the presence of cholesterol may greatly facilitate peptide insertion into the bilayer. The preferential interaction of amyloidogenic proteins with lipid raft domains ensures that cholesterol is indeed present underneath the sphingolipids that have attracted the unstructured monomer (Fig. 6). Once inserted in the membrane, the amyloidogenic protein will immediately find cholesterol and interact with its free side – that is, the face that is not in contact with sphingolipids (for a detailed explanation of cholesterol topology, see Ref. Reference Fantini and Barrantes18).
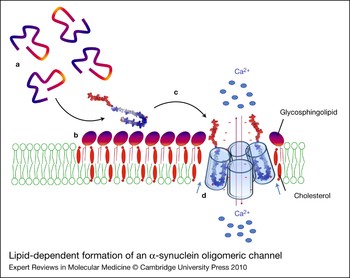
Figure 6. Lipid-dependent formation of an α-synuclein oligomeric channel. Unfolded α-synuclein monomers (a) are attracted by the polar heads of glycosphingolipids and concentrated on the surface of sphingolipid–cholesterol membrane domains (b). Sphingolipids, complexed with cholesterol, induce a conformational change (unstructured to α-helix transition) of α-synuclein. The newly formed α-helices can insert into the plasma membrane (c) where they can further oligomerise under the control of cholesterol molecules (blue arrows) (d) and eventually form an oligomeric ion channel. Such channels are thought to disturb membrane permeability to calcium ions (red arrows), resulting in neuronal dysfunction and toxicity. The minus signs (in red) refer to the negative charges of α-synuclein.
How cholesterol could regulate the formation of oligomeric pores and channels
By stimulating the oligomerisation of membrane-inserted amyloidogenic proteins, cholesterol could facilitate the formation of a pore-like assembly displaying ion channel properties (Fig. 1). Quist et al. (Ref. Reference Quist10) have hypothesised that amyloid pore/channels provide the most direct pathway for inducing neurodegenerative effects, through loss of ionic homeostasis increasing cell calcium to toxic levels. The structure of amyloid pores, which have been observed by atomic force microscopy (Refs Reference Lashuel9, Reference Quist10), remains to be experimentally elucidated at the atomic level. Computational models have suggested that most amyloid pores are probably formed by a complex assembly of β-rich protofibrils or oligomers (Ref. Reference Butterfield and Lashuel130), as shown in Figure 1. However, in the case of α-synuclein, Tsigelny et al. (Ref. Reference Tsigelny101) have elegantly modelled a realistic oligomeric channel formed by the assembly of α-helical monomers. The possible events leading to the formation of an α-synuclein oligomeric channel under the dual control of sphingolipids and cholesterol are summarised in Figure 6. There is a striking similarity between α-synuclein (Refs Reference Zakharov69, Reference Di Pasquale31) and colicin E1, a bacterial toxin that also inserts into anionic areas of the plasma membrane, forming channel-like structures consisting of an α-helix bundle (Ref. Reference Stroud102). Moreover, it has been hypothesised that amyloid pores might be similar to β-barrel pore-forming bacterial toxins (Ref. Reference Lashuel and Lansbury103). Since cholesterol controls the oligomerisation and insertion of these bacterial toxins (Ref. Reference Palmer104) in the target membrane, it is likely that it is also required for the assembly of amyloid pores.
How could cholesterol control the oligomerisation process of membrane-inserted amyloidogenic proteins and facilitate the formation of amyloid pores? The recent high-resolution crystal structure of an engineered human adrenergic β2 receptor, suggesting a molecular mechanism by which cholesterol mediates receptor dimerisation (Ref. Reference Cherezov105), might give some clues to this fundamental issue. On the basis of these structural data, one could hypothesise that cholesterol binds to membrane-embedded fragments of amyloidogenic proteins, facilitates their recruitment and coordinates their oligomerisation. Overall, this would mean that amyloid pore formation and ion channel function are under the control of both sphingolipids and cholesterol, in agreement with recent data obtained in model membranes with α-synuclein (Ref. Reference Di Pasquale31) and amylin (Ref. Reference Cho, Trikha and Jeremic106). Finally, it has been reported that spherical amyloid oligomers or protofibrils can also increase membrane conductance without forming ion channels or pores (Ref. Reference Kayed107). It is not known if cholesterol and/or sphingolipids are involved in this nonspecific permeabilising activity.
Changes in lipid content of the brain: what impact on neurodegenerative diseases?
The involvement of gangliosides in several neurodegenerative diseases is not totally surprising if one considers that these sphingolipids are critical for neuronal integrity and plasticity (Ref. Reference Mocchetti108) and synaptic function (Ref. Reference Thomas and Brewer109). Since the probability of acquiring these diseases gradually increases with age, it is important to evaluate how ganglioside expression varies during the lifetime of individuals, and whether these diseases can alter this process (Ref. Reference Posse de Chaves and Sipione110). In vitro, the differentiation of PC12 cells into neuron-like cells caused a marked increase in both gangliosides and cholesterol, and thereby greatly potentiated the accumulation and cytotoxicity of Aβ (Ref. Reference Wakabayashi and Matsuzaki111). GM1 seems to control the oligomerisation and aggregation process of Aβ and α-synuclein (Refs Reference Yanagisawa29, Reference Martinez30, Reference Di Pasquale31). GM3 has been shown to control α-synuclein channel formation and to correct the channelopathy induced in planar lipid membranes by the Parkinson-disease-linked E46K mutant of the protein (Ref. Reference Di Pasquale31). In the human brain, GM3 is a minor ganglioside, which, in marked contrast with the major ganglioside species GM1 and GD1A, shows a regular increase with age (Ref. Reference Svennerholm112). The expression of GM3 is highly regulated during brain development (Ref. Reference Yu, Nakatani and Yanagisawa113), and this ganglioside has been implicated in the regulation of neuronal cell death (Ref. Reference Sohn114). Moreover, the homozygous loss-of-function mutation of GM3 synthase, which totally suppresses the expression of GM3 and all GM3-derived gangliosides, has been linked to an infantile symptomatic epilepsy syndrome (Ref. Reference Simpson115). Taken together, these data indicate that ganglioside GM3 probably plays a more important role in brain physiology and pathology than its low expression levels in adult brain could have suggested in the past.
Several studies support the view that deregulation of lipid metabolism is an important feature of neurodegenerative diseases. The three main lipid categories – glycerophospholipids, sphingolipids and cholesterol – are affected (Refs Reference Svennerholm and Gottfries116, Reference Grimm117, Reference Bandaru118, Reference Ariga, McDonald and Yu119, Reference He120, Reference Xu and Huang121). A large body of data has established a link between cholesterol homeostasis, apolipoprotein E polymorphism and APP processing (Ref. Reference Chauhan122). Correspondingly, cholesterol-lowering strategies with statins have acquired potential therapeutic importance in treating Alzheimer disease (Ref. Reference Kandiah and Feldman123), although success has been variable (Ref. Reference Rea124). Statins have also proved to be efficient in decreasing α-synuclein aggregation and neuronal toxicity in animal models of Parkinson disease (Ref. Reference Koob98). Specific changes in ganglioside content have been detected in the brains of patients with Alzheimer disease (Refs Reference Svennerholm and Gottfries116, Reference Kracun125): a decrease or even loss of the major gangliosides GM1, GD1a, GD1b and GT1b, and an increase in GM2, GM3 and GD3. Similar alterations of ganglioside expression were observed in the brains of transgenic mouse models of Alzheimer disease (Ref. Reference Barrier126). Regional variations in ganglioside content have been shown in Alzheimer brains (Ref. Reference Molander-Melin127). Moreover, the balance between NFA and HFA ceramides is altered in animal models (Ref. Reference Barrier128). Interestingly, the authors of the latter study showed that there is a gender-dependent accumulation of ceramides in the cerebral cortex: female mice exhibited a strong increase in HFA species, and males in NFA species. This observation could be linked to the increased risk of women versus men for developing Alzheimer disease (Ref. Reference Fratiglioni129). Because cholesterol can exert distinct effects on Aβ–membrane interactions according to the NFA:HFA content of brain sphingolipids (Ref. Reference Yahi, Aulas and Fantini52), it will be interesting to determine how cholesterol impacts on Aβ–glycosphingolipid interaction in men and women suffering from Alzheimer disease.
Perspectives
How amyloidogenic proteins kill neurons is still a mystery. In particular, as recently discussed by Butterfield and Lashuel (Ref. Reference Butterfield and Lashuel130), ‘there remains a knowledge gap regarding the molecular-level details by which amyloid-forming proteins act on the membrane and induce membrane permeabilization’. Deciphering the complex interplay between amyloidogenic proteins and membrane lipids, especially sphingolipids and cholesterol, will certainly help to achieve a clearer view on this enigma. Several milestones have already been reached, which have inspired an important part of today's research efforts worldwide. Despite their lack of amino acid sequence homology, amyloidogenic proteins share a number of intriguing properties: (1) an important conformational plasticity, allowing the same protein to remain unordered or to form highly ordered α and β structures (Refs Reference Uversky4, Reference Uversky5); (2) self-oligomerising capacities that revealed unexpected common antigenic properties (Ref. Reference Kayed7); (3) the ability to form pore-like structures with ion channel properties (Refs Reference Lashuel9, Reference Quist10); (4) self-aggregating activity leading to fibrillation and plaque deposition (Ref. Reference Iversen131); and (5) common structural motifs allowing specific interactions with sphingolipids (Ref. Reference Fantini49) and cholesterol (yet uncharacterised) in lipid raft domains of the plasma membrane. It is intriguing that HIV-1, which interacts with glycosphingolipids through the V3 domain of its surface glycoprotein gp120, can induce major neurological disabilities, identified as HIV-1-associated dementia (Ref. Reference Posse de Chaves and Sipione110). In this respect, it is also worth noting that the protein CLN3, involved in Batten disease (the juvenile form of neuronal ceroid lipofuscinosis), also binds to GalCer through a V3-like sphingolipid-binding domain (Ref. Reference Persaud-Sawin132). This neural disease is caused by a mutation in this domain, namely E295K (Ref. Reference Zhong133), which is analogous to mutations E22K in Aβ, E46K in α-synuclein and E200K in PrP, all associated with inherited forms of neurodegenerative disease and located in the sphingolipid-binding domains of these proteins. At least for two of these proteins (PrP and α-synuclein), the substitution of an anionic glutamic acid side chain by a cationic lysine resulted in altered binding to membrane sphingolipids (Refs Reference Mahfoud27, Reference Di Pasquale31). This further emphasises the key role of the sphingolipid-binding domain in neuronal diseases.
Future studies will be necessary to better evaluate the neurotoxicity of the various forms of amyloidogenic proteins, including monomers, oligomers and aggregates, and to understand how these different species cooperate to accelerate (or slow down) the onset of neurodegenerative symptoms. This will help to decide on a rational basis which therapeutic strategy should be used at the different stages of the diseases. This will be possible with the finding of nontoxic drugs specifically affecting amyloid channels (Ref. Reference Diaz134) or amyloid aggregation (Ref. Reference Panza135). Combining molecular (Refs Reference Di Pasquale31, Reference Yahi, Aulas and Fantini52), biophysical (Refs Reference Munishkina and Fink14, Reference Di Pasquale31, Reference Zakharov69, Reference Waschuk136, Reference Thakur, Micic and Leblanc137), cellular (Refs Reference Arispe and Doh93, Reference Sohn114, Reference Furukawa138) and animal (Refs Reference Barrier126, Reference Barrier128, Reference Oikawa139) studies will allow a better characterisation of the sphingolipids that regulate amyloid oligomerisation/aggregation. Biophysical properties of the membrane such as membrane curvature should be taken into consideration (Ref. Reference Butterfield and Lashuel130). Indeed, depending on the surface curvature of the model membrane, membrane-bound α-synuclein can adopt an extended helix (Ref. Reference Georgieva140), a bent helix (Ref. Reference Jao141) or an antiparallel helix–turn–helix conformation (Ref. Reference Ulmer142) (for a review see Ref. Reference Butterfield and Lashuel130). Interestingly, GM1 and GM3 are concentrated in membrane areas that markedly differ in membrane curvature (Ref. Reference Chen, Qin and Chen143), so that on binding to distinct gangliosides, the same amyloid protein could adopt distinct conformations. This could be the case for α-synuclein, which recognises both GM1 (Ref. Reference Martinez30) and GM3 (Ref. Reference Di Pasquale31). Antiganglioside antibodies could interfere with normal ganglioside function and could thus play a role in disease pathogenesis, as anti-GM1 antibodies probably do in some Parkinson patients (Refs Reference Posse de Chaves and Sipione110, Reference Zappia144). Careful determinations of the titres of these antibodies in various biological fluids, including cerebrospinal fluid, will be particularly informative. Beneficial effects of ganglioside supplementation have been reported in animal models of Parkinson disease (Ref. Reference Sautter145). This warrants further investigation. Enzymes involved in glycosphingolipid metabolism might represent targets that inhibit both the production and amyloid aggregation of Alzheimer Aβ peptides (Ref. Reference Tamboli146). Correspondingly, lipid raft disruption has been shown to protect neurons against amyloid oligomer toxicity in vitro (Ref. Reference Malchiodi-Albedi147). We need to better understand how cholesterol interacts with amyloidogenic proteins, regulates their supramolecular structures and is involved in the pathophysiology of neurodegenerative diseases (Refs Reference Hu86, Reference Liu87). We should also determine how mutations of amyloidogenic proteins affect their interaction with neural membranes (Refs Reference Mahfoud27, Reference Zakharov69, Reference Bodner148). A decisive breakthrough will be to understand why neurodegenerative diseases involve specific brain areas, and how local lipid composition may account for such geographic restrictions (Refs Reference Molander-Melin127, Reference Panza135). Finally, we have to identify the environmental molecules (such as food contaminants, pesticides and mycotoxins) that could modulate amyloid formation through direct binding to amyloid proteins and could represent important risk factors for neurodegenerative diseases (Ref. Reference Uversky149).
Acknowledgements and funding
The authors thank the peer reviewers for their interest in this work and their very constructive and thoughtful comments, which have stimulated us to improve our manuscript.







 Open access
Open access