


Metabolic syndrome is generally defined as a cluster of risk factors for cardiovascular disease and type 2 diabetes mellitus (T2DM) including central obesity, arterial hypertension, dyslipidaemia and elevated fasting glucose (Ref. Reference Alberti1). Impaired glucose homeostasis, as observed in patients with metabolic syndrome, frequently progresses to overt T2DM, which in 2010 affected 344 million patients worldwide (Ref. 2). Hyperglycaemia in diabetic patients can lead to life-threatening complications such as coronary heart disease, stroke and nonalcoholic fatty liver disease (Refs Reference Giacco and Brownlee3, Reference Roden4, Reference Chiasson5).
Strict control of the level of circulating glucose within a narrow physiological range supplies sufficient energy for organs and avoids hyperglycaemia. Glucose homeostasis is largely maintained by the insulin–glucagon system, which compensates for physiological fluctuations in blood glucose caused by food intake and physical activity, or by stress conditions such as hypoxia and inflammation. Insulin and glucagon are released from β- and α-cells, respectively, in the endocrine part of the pancreas. Insulin lowers blood glucose by stimulating glucose uptake and storage (glycogen synthesis and lipogenesis) in skeletal muscle and adipose tissue. In the liver, insulin blocks the release and neogenesis of glucose and stimulates glucose storage. In addition, insulin stimulates protein synthesis, regulates mitochondrial biogenesis and blocks autophagy. Glucagon antagonises the action of insulin, mostly in the liver, where it stimulates gluconeogenesis and thereby increases blood glucose level. The secretion of insulin and glucagon is regulated in a reciprocal manner, which avoids glycaemic volatility because of their opposing effects. It was proposed that the glucose-induced secretion of insulin inhibits glucagon secretion from α-cells in a paracrine manner (Ref. Reference Unger and Orci6). Furthermore, incretin hormones [e.g. glucagon-like peptide 1 (GLP-1)] secreted postprandially by the gut potentiate glucose-mediated insulin secretion and block glucagon secretion (Ref. Reference Holst, Vilsboll and Deacon7). In addition, physiological conditions such as low intracellular energy level and cellular stress affect whole-body glucose homeostasis by interfering with insulin action.
Signal transduction from a stimulus to the regulation of cellular processes, including those involved in glucose homeostasis, is primarily dependent on protein kinase signalling. On activation, protein kinases determine the output of metabolic processes by transcriptional and post-translational regulation of rate-limiting enzymes, such as glycogen synthase 1 (GYS1) and fatty acid synthase (FASN, FAS). The insulin receptor (INSR, IR) activates various downstream pathways that control energy homeostasis, including phosphoinositide-3-kinase (PI3K)/v-akt murine thymoma viral oncogene homologue [AKT, also known as protein kinase B (PKB)] and the mitogen-activated protein kinase 3/1 (MAPK3/1, ERK1/2). Whereas the PI3K/AKT pathway is considered to be the major effector of metabolic insulin action, insulin-independent kinases also contribute to metabolic control. AMP-activated protein kinase (AMPK) is mostly activated by low intracellular energy levels and inhibits anabolic processes, stimulates energy-producing catabolic processes and lowers blood glucose level. Because correct functioning of the PI3K/AKT, MAPK and AMPK pathways is essential for proper metabolic control and their dysfunction often leads to impaired glucose homeostasis, these pathways are attractive therapeutic targets (Refs Reference Cho8, Reference Sabio and Davis9, Reference Viollet10). However, PI3K/AKT, MAPK and AMPK are also involved in several other fundamental cellular processes, including cell proliferation and survival, and thus global therapeutic modification of their activities could induce severe side effects.
Today, specific kinase inhibitors are used successfully for immunosuppression and in the treatment of inflammatory disease and diverse cancer types. However, because proper activation of the PI3K/AKT pathway is required for insulin action, kinase inhibitors targeting PI3K/AKT and downstream effectors might impair metabolic control. Even though inappropriate activation of MAPKs, especially of c-Jun N-terminal kinase (MAPK8, JNK), is considered to have a critical role in acquired insulin resistance, no therapies based on MAPKs are available so far. The only drug targeting protein kinase activity that is widely used today in the treatment of insulin resistance and diabetes is metformin, which is thought to operate mainly by activating AMPK. Although our understanding of the role of protein kinases in the regulation of glucose homeostasis has increased significantly during the past decade, only limited translation into therapies against the metabolic syndrome has occurred. The purpose of this present review is to summarise the signal transduction mechanisms involving PI3K/AKT, MAPK and AMPK with respect to their role in glucose homeostasis and to discuss current clinical implications.
The PI3K–AKT signalling pathway is the major effector of metabolic insulin action
Insulin is an indispensable regulator of glucose homeostasis, and T2DM is characterised by postreceptor insulin resistance combined with β-cell failure. Insulin signalling is initiated by the binding of insulin to the extracellular α-subunits of the heterotetrameric IR. This interaction induces conformational changes and facilitates autophosphorylation of tyrosine residues on the intracellular part of membrane-spanning β-subunits. These phosphotyrosines then attract a family of adaptor molecules, the insulin receptor substrates (IRSs). On interaction with the IR, IRS proteins themselves are tyrosine phosphorylated, which is partially mediated by the tyrosine kinase activity of the IR and also by other kinases. Once phosphorylated, IRS proteins attract downstream signalling molecules, thereby linking the activated IR to the various downstream signalling pathways (Ref. Reference White11).
Molecular mechanism of the PI3K/AKT signalling pathway downstream of insulin
The PI3K/AKT pathway is required for insulin-dependent regulation of systemic and cellular metabolism (Ref. Reference Cho8). Besides insulin, many other growth factors, cytokines and environmental stresses can activate PI3K/AKT, mainly in the regulation of cell proliferation, motility, differentiation and survival (Ref. Reference Zhuravleva, Tschopp, Hemmings, Dufour and Clavien12). Thus, PI3K/AKT action is highly context dependent, which is at least partially mediated by the recruitment of different isoforms of PI3K (including p85α, p110α, p110β) and AKT (AKT1, AKT2, AKT3) downstream of individual stimuli (Refs Reference Vanhaesebroeck13, Reference Manning and Cantley14, Reference Schultze15). The AKT isoforms are encoded by individual genes located on different chromosomes, share approximately 80% identity in their amino acid sequences and form the same protein structure, including an N-terminal pleckstrin homology (PH), a catalytic domain and a C-terminal regulatory domain (Ref. Reference Hanada, Feng and Hemmings16). Among the AKT isoforms, AKT2 is considered to be the major isoform required for metabolic insulin action. Although intensively investigated, the exact molecular mechanisms underlying isoform and context specificity are still not fully elucidated. Here we focus on the function of the PI3K/AKT pathway downstream of IR and IRS proteins and its role in glucose homeostasis.
The PI3K/AKT pathway is activated downstream of the IR by binding an SH2 domain within the regulatory subunit of PI3K (p85) to phosphotyrosines in IRS1/2. This leads to recruitment and activation of the catalytic subunit of PI3K (p110). Once activated, PI3K converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3) at the plasma membrane. AKT binds through its PH domain to PIP3, which facilitates activation of AKT by upstream kinases. Initially, 3-phosphoinositide-dependent protein kinase-1 (PDPK1, PDK1) induces about 10% of kinase activity by phosphorylating Thr308 in the catalytic domain of AKT. Subsequently, mammalian target of rapamycin complex 2 (mTORC2), DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated kinase (ATM) induce full kinase activity of AKT by phosphorylating Ser473 in the regulatory domain. Although DNA-PK can phosphorylate AKT at Ser473 on insulin stimulation in vitro, it is thought to activate AKT in vivo mainly following stress such as DNA damage (Refs Reference Feng17, Reference Bozulic and Hemmings18, Reference Surucu19). mTORC2 is considered to be the predominant AKT Ser473 kinase downstream of insulin and growth factor stimuli (Ref. Reference Bozulic and Hemmings18). On activation, AKT is released from the plasma membrane and translocates to cellular compartments, such as the cytoplasm, mitochondria and nucleus, where it phosphorylates its many substrates. Substrates implicated in the regulation of cellular metabolism include glycogen synthase kinase 3β (GSK3β), forkhead box protein O1 (FOXO1) and AKT substrate 160 (TBC1D4, AS160), which regulate glycogen synthesis, gluconeogenesis and glucose uptake, respectively. AKT also activates mTORC1 by inhibiting tuberous sclerosis complex 1/2 (TSC1/2). Activated mTORC1 upregulates mitochondrial biogenesis, inhibits autophagy and induces protein synthesis by regulation of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), unc-51-like kinase 1 (ULK1), and ribosomal protein S6 kinase (S6K) and eIF4E-binding protein 1 (4E-BP1), respectively. PDK1 also activates isoforms of protein kinase C (PKCλ/ζ), which are required for Glut4-dependent regulation of glucose uptake. Moreover, AKT and PKCλ/ζ control de novo lipogenesis by regulating lipogenic genes, such as sterol regulatory element-binding transcription factor 1 (SREBF1, SREBP1c) and peroxisome proliferator-activated receptor γ (PPARγ) (Refs Reference Leavens20, Reference Taniguchi21). The mechanisms by which AKT and PKCλ/ζ regulate lipogenic genes are not yet completely understood.
The insulin–PI3K/AKT pathway is negatively regulated at different levels. Phosphatases, including protein tyrosine phosphatase nonreceptor type 1 (PTPN1, PTP1B), phosphatase and tensin homologue (PTEN) and protein phosphatase 2A (PP2A), dephosphorylate and thereby inhibit IR, IRS1/2, PIP3 and AKT, respectively. AKT activity can also be inhibited by binding partners, such as thioesterase superfamily member 4 (THEM4, CTMP) and tribbles homologue 3 Drosophila (TRIB3) (Refs Reference Maira22, Reference Du23). Whereas the function of most AKT-binding partners in glucose homeostasis remains to be elucidated, TRIB3 was shown to inhibit insulin signalling (Ref. Reference Du23). Furthermore, negative-feedback loops are implemented in the PI3K/AKT pathway that downregulate insulin signalling. GSK3β, mTORC1 and S6K can phosphorylate IRS on serine residues, which can lead to their ubiquitylation and proteolytic breakdown (reviewed in Refs Reference Zhuravleva, Tschopp, Hemmings, Dufour and Clavien12, Reference Brazil and Hemmings24, Reference Bhaskar and Hay25) (Fig. 1).
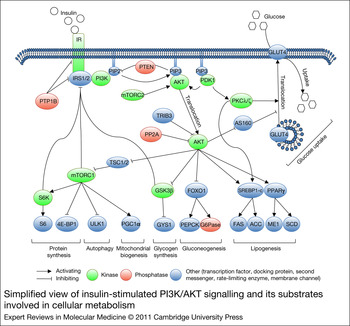
Figure 1. Simplified view of insulin-stimulated PI3K/AKT signalling and its substrates involved in cellular metabolism. A detailed description is given in the text. Abbreviations: ACACA, ACC, acetyl-CoA carboxylase; AKT, v-akt murine thymoma viral oncogene homologue 1; 4E-BP1, eIF4E-binding protein 1; FOXO1, forkhead box O1; G6Pase, glucose-6-phosphatase; GSK3β, glycogen synthase kinase 3β; GYS1, glycogen synthase; INSR, IR, insulin receptor; IRS1/2, insulin receptor substrates 1/2; ME1, malic enzyme 1; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mTOR complex 2; PDPK1, PDK1, 3-phosphoinositide-dependent protein kinase-1; PGC1α, peroxisome proliferator-activated receptor gamma, coactivator 1α; PI3K, phosphoinositide-3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate; PKC1, PEPCK, phosphoenolpyruvate carboxykinase 1; PKCλ/ζ, protein kinase Cλ/ζ; PPARγ, peroxisome proliferator-activated receptor g; PP2A, protein phosphatase 2A; PTPN1, PTP1b, protein tyrosine phosphatase, non-receptor type 1; RPS6, S6, ribosomal protein S6; SCD, stearoyl-CoA desaturase; S6K, ribosomal protein S6 kinase; SLC2A4, GLUT4, solute carrier family 2; SREBF1, SREBP1-c, sterol regulatory element binding transcription factor 1; TBC1D4, AS160, AKT substrate 160; TRIB3, tribbles homologue 3; TSC1/2, tuberous sclerosis complex 1/2; ULK1, unc-51-like kinase 1.
Genetic alterations in components of the insulin signalling pathway can impair or improve metabolic control
Many studies have been carried out in mice and humans and have been pivotal in defining the molecular events underlying insulin signalling. Most patients develop insulin resistance and T2DM as a result of polygenetic predisposition in combination with overnutrition and obesity (acquired insulin resistance). Monogenetic defects causing diabetes account for only 1–5% of cases and have been found in loci encoding elements of the insulin signalling pathway, transcription factors and rate-limiting enzymes of glucose metabolism (e.g. hepatocyte nuclear factor 4α and glucokinase) and also in mitochondrial genes (Ref. Reference Permutt, Wasson and Cox26). Interestingly, both enhancement of insulin signalling by deletion of negative regulators and specific interference with its action by deleting targets normally activated by insulin can improve metabolic control and protect against diabetes in mice.
From IR to AKT: genetic mutations and their effects on insulin sensitivity in humans and mice
Patients with loss-of-function mutations in IR are severely insulin resistant and display signs of hyperglycaemia and hyperinsulinaemia, thus indicating that IR is essential for insulin action (Refs Reference Kahn27, Reference Odawara28, Reference Musso29). Experiments in vitro have confirmed that amino acid substitutions in the tyrosine kinase domain of IR found in patients, such as glycine (G) to valine (V) at position 996 (G996V) and Q1131R, indeed block insulin signalling, as shown by markedly reduced IR tyrosine kinase activity and diminished phosphorylation of IRS1/2 (Refs Reference Odawara28, Reference Kishimoto30).
Of the postreceptor gene mutations in the insulin signalling cascade, only a few were found to cause severe insulin resistance in humans. Several variants of IRS1 and IRS2 have been identified in patients with insulin resistance. Two IRS1 variants, a common (G972R) and a rare (T608R) polymorphism, were associated with reduced insulin sensitivity in obese men and severe insulin resistance, respectively (Refs Reference Clausen31, Reference Esposito32). Both polymorphisms are located in regions implicated in PI3K binding and abolished insulin-stimulated PI3K activity in cell culture models (Refs Reference Esposito32, Reference Almind33). By contrast, variants of IRS2 were not associated with insulin resistance, and their biochemical properties were not characterised (Refs Reference Bottomley34, Reference Bernal35). Of the known polymorphisms in p85α and p110β subunits of PI3K, only an R409Q amino acid substitution in p85α was shown to compromise insulin-stimulated PI3K activity (Refs Reference Kossila36, Reference Baynes37). Remarkably, a mutation identified in AKT2 resulting in an R274H amino acid substitution in the kinase domain was associated with autosomal dominant inherited severe insulin resistance. AKT2 R274H has greatly reduced kinase activity and acts in a dominant-negative manner in that its overexpression blocks the inhibition of FOXA2 in HepG2 cells and impairs adipocyte differentiation in vitro (Ref. Reference George38).
Findings in transgenic mice complement the above observations. Mice deficient in the IR develop severe hyperglycaemia within hours after birth and die within days as a result of severe ketoacidosis (Refs Reference Accili39, Reference Joshi40). IRS1-deficient mice have peripheral insulin resistance, but show only slight hyperglycaemia because of compensatory hyperinsulinaemia (Refs Reference Araki41, Reference Tamemoto42). A more severe metabolic phenotype was observed in mice deficient in IRS2. These animals are also insulin resistant, but show hyperglycaemia as a result of impaired adaptation of β-cell mass (Ref. Reference Withers43). By contrast, specific loss of elements of insulin signalling can also improve metabolic control. It was shown that mice with an adipose-tissue-specific deletion of Ir are protected against obesity and obesity-related insulin resistance (Ref. Reference Blüher44). Whereas p85α R409Q is associated with reduced insulin sensitivity in humans, loss of p85α by mutation of the corresponding gene Pik3r1 (which encodes p85α, p55α and p50α) resulted in improved glucose tolerance and hypoglycaemia in mice (Refs Reference Terauchi45, Reference Fruman46). It was suggested that the loss of p85α is compensated by p50α, which generated an increase in PIP3 on insulin stimulation (Ref. Reference Terauchi45). However, there may be further compensatory mechanisms, given that mice lacking all three isoforms of Pik3r1 are also hypoglycaemic (Ref. Reference Fruman46). These studies demonstrate that ablation of proteins can have effects different from loss-of-function mutations and also from inhibitor treatments in which the inoperative protein remains present. For detailed descriptions of these mouse models, the reader is referred to another review (Ref. Reference Lee and Cox47).
As described above, loss-of-function mutations in genes of the insulin signalling pathway mostly reduce insulin sensitivity to varying degrees. These findings support the notion that these genes are required for insulin action and are the basis of our understanding of the molecular mechanisms underlying insulin signalling and the development of diabetes. Thus, at first sight, it appears desirable to enhance insulin signalling in order to counteract the development of diabetes. However, the observation that adipose-tissue-specific IR deficiency can improve metabolic control and protect against obesity suggests an interesting alternative.
Diverse effects on glucose homeostasis are observed after deletion of individual AKT isoforms and downstream protein kinases in mice
The mechanisms of insulin signalling at the level of and downstream of, AKT isoforms have been studied extensively in transgenic mouse models. AKT1 and AKT2 are ubiquitously expressed, with high levels in classical insulin target tissues such as the liver, skeletal muscle and adipose tissue (Refs Reference Buzzi48, Reference Yang49). By contrast, the expression of AKT3 appears more restricted and is found mainly in the brain, the testis, adipose tissue and pancreatic islets (Refs Reference Buzzi48, Reference Yang49). As in the case of humans, mice lacking AKT2 are insulin resistant, hyperglycaemic and hyperinsulineamic (Refs Reference Cho8, Reference Buzzi48, Reference Garofalo50). Deficiency in AKT3 does not result in metabolic aberrations. However, somewhat conflicting results have been obtained with mice deficient in AKT1. Two studies reported no role for AKT1; however, a third study described higher insulin sensitivity and improved metabolic control (Refs Reference Buzzi48, Reference Cho51, Reference Chen52). The molecular mechanisms underlying this improved insulin sensitivity in AKT1-deficient mice have not been defined. Although highly similar in structure, loss of individual AKT isoforms results in distinct phenotypes, indicating that AKT isoforms exert nonredundant functions. This can be partially explained by divergent expression patterns, but we are far from understanding the molecular mechanisms underlying specificity (Ref. Reference Schultze15).
The results obtained in mouse models indicate that individual downstream effectors of AKT exert distinct and tissue-specific functions. GSK3β inhibits glycogen synthesis by phosphorylating GYS1 and is negatively regulated by AKT. Accordingly, mice with specific deletion of Gsk3b in skeletal muscle but not in the liver showed improved glucose tolerance owing to enhanced GYS1 activity and glycogen deposition (Ref. Reference Patel53). Additionally, it was shown that mice with a pancreatic β-cell-specific deletion of Gsk3b display increased β-cell mass and improved glucose tolerance and are protected against genetically and diet-induced diabetes. This increase in β-cell mass might occur as a result of loss of GSK3β-mediated feedback inhibition of insulin signalling, which is known to increase β-cell proliferation (Refs Reference Liu54, Reference Tanabe55).
mTORC1 and its downstream target S6K are indirectly activated by AKT2, and their roles have also been studied in mice. Activation of mTORC1 in β-cells by deletion of Tsc1 or Tsc2 increased cell size, proliferation and insulin production. Thus, β-cell-specific activation of mTORC1 improved glucose-stimulated insulin secretion and glucose tolerance in mice (Refs Reference Mori56, Reference Rachdi57). Conversely, mice with a whole-body S6K deficiency showed reduced β-cell mass and hypoinsulinaemia (Ref. Reference Pende58). Ablation of mTORC1 activity in skeletal muscle in mice by deletion of Raptor reduced oxidative capacity by the downregulation of genes involved in mitochondrial biogenesis. Moreover, the glycogen content of the muscle in these mice was increased, most likely because of enhanced inhibition of GSK3β by hyperactivated AKT. As a result, these mice suffered from progressive muscle dystrophy and were glucose intolerant (Ref. Reference Bentzinger59). Interestingly, mice with an adipocyte-specific mTORC1 deficiency as well as those with a whole-body S6K deficiency were protected against diet-induced obesity and insulin resistance. The authors proposed that the protective effects are based on increased energy expenditure and enhanced insulin signalling, which are probably due to loss of negative feedback regulation in adipose tissue (Refs Reference Polak60, Reference Um61). Recently, it was shown that mice with liver-specific activation of mTORC1 by deletion of Tsc1 are glucose intolerant but, are protected against diet-induced hepatic steatosis. The authors also showed that inhibition of mTOR by rapamycin does not reduce hepatic lipid accumulation in mice fed a high-fat diet. Thus, it was concluded that mTORC1 is not required and not sufficient to increase hepatic lipids, but rather protects against diet-induced hepatic steatosis by enhancing fat utilisation and gluconeogenesis in the liver (Ref. Reference Kenerson, Yeh and Yeung62) (Table 1).
Table 1. Overview of mouse models for AKT isoforms and downstream targets

Further descriptions are given in the text. NR, not reported; UC, unchanged; +, improved; −, reduced; (−/−), homozygous mutant; (+/−), heterozygous mutant.
These findings show that not only AKT isoforms but also their downstream effectors perform distinct functions in the regulation of glucose homeostasis. Moreover, the impact on metabolic control of modulating the activity of downstream components in the insulin signalling cascade largely depends on the targeted tissue, as demonstrated in the case of mTORC1 and S6K. Thus, the development of techniques for tissue-specific, but not systemic, targeting of downstream components could allow further adaption of current therapies to individual demands, such as improving β-cell function, reducing hepatic lipid content and restoring insulin response in skeletal muscle.
Improved glucose homeostasis in mice lacking negative regulators of PI3K/AKT
As mentioned above, phosphatases such as PTP1B and PTEN antagonise insulin signalling. PTP1B downregulates insulin-stimulated PI3K/AKT signalling by dephosphorylating IR and IRS1/2 in a more specific manner than PTEN, which inhibits PI3K/AKT signalling by dephosphorylating PIP3. Because several other growth factors, such as EGF and PDGF, can also increase levels of PIP3 by stimulating PI3K, PTEN appears to be a critical antagonist of all PI3K-dependent AKT stimuli. Notably, both PTP1B deficiency and Pten hemizygosity result in improved glucose tolerance and insulin sensitivity in mice (Refs Reference Elchebly63, Reference Wong64). Similar phenotypes were found in mice with tissue-specific PTP1B deficiency in muscle or liver, and PTEN deficiency in muscle, adipose tissue or liver (Refs Reference Delibegovic65, Reference Delibegovic66, Reference Wijesekara67, Reference Kurlawalla-Martinez68, Reference Horie69, Reference Stiles70). Furthermore, it was shown that mice with whole-body and muscle-specific PTP1B deficiency, and mice lacking PTEN in muscle and pancreas, are protected against diet-induced insulin resistance (Refs Reference Elchebly63, Reference Delibegovic65, Reference Wijesekara67, Reference Tong71). In contrast to PTP1B-deficient mice, mice with Pten hemizygosity and mice with PTEN deficiency in hepatocytes develop tumours in various organs or progressive hepatic steatosis with the development of liver cancer, respectively (Refs Reference Horie69, Reference Stiles70, Reference Chen72) (Table 2). These phenotypes indicate that PTEN is required to control growth-factor-stimulated PI3K/AKT signalling. Moreover, PTEN was shown to have a phosphatase-independent tumour-suppressive function in the nucleus, which might also have a role in tumour development in mice (Ref. Reference Song73).
Table 2. Overview of mouse models for the role of Pten and Ptp1b in glucose homeostasis

UC, unchanged; +, improved; −, reduced; (−/−), homozygous mutant; (+/−), hemizygous.
Recent evidence suggests that the targeting of negative regulators further downstream, such as TRIB3, might enhance insulin signalling without global activation of the PI3K/AKT pathway. Whereas mice with whole-body TRIB3 deficiency showed no alterations in metabolic control under normal conditions, TRIB3 was shown to be upregulated in the liver of diabetic mice and hepatic overexpression of TRIB3 impaired glucose tolerance (Refs Reference Du23, Reference Okamoto74, Reference Koo75). Because TRIB3 seems to be dispensable under normal conditions, but seems to contribute to obesity-induced insulin resistance, it might represent an attractive therapeutic target.
As underlined by the complex phenotype of PTEN-deficient mice, the inhibition of negative regulators can lead to global activation of PI3K/AKT with severe side effects such as hepatic steatosis and cancer. Thus, the safe targeting of negative regulators of insulin signalling may be out of reach until the regulation of context-specific stimulation is understood. The targeting of negative regulators further downstream, such as TRIB3, could be more specific and have improved side-effect profiles.
mTOR inhibitors in clinical use and how they affect glucose homeostasis
Although results from the studies described above show that interfering with PI3K/AKT/mTOR signalling mostly leads to insulin resistance, its inhibition is an attractive treatment option for various other diseases. Inhibition of PI3K/AKT/mTOR signalling should be considered especially in cancer therapy, because inappropriate activation of this pathway is frequently observed in many tumour types. Indeed, the mTOR inhibitors temsirolimus and everolimus have been approved for the treatment of metastatic renal cell carcinoma (mRCC) and improve overall or progression-free survival (Refs Reference Motzer76, Reference Hudes77). Current trials explore the efficiency of mTOR inhibitors when used in combination with other therapies, including small-molecule tyrosine kinase inhibitors or VEGF-directed antibodies (Ref. Reference Pal and Figlin78). In addition to mRCC, an increasing number of clinical trials study the effects of mTOR inhibition in other diseases, such as pancreatic neuroendocrine tumours, astrocytomas, lymphangioleiomyomatosis and autosomal dominant polycystic kidney disease (Refs Reference Bissler79, Reference Qian80, Reference Torres81, Reference Yao82, Reference Krueger83, Reference Witzig84). Owing to their inhibitory effect on proliferation of lymphocytes, both compounds have also been used for immunosuppression after transplantation.
However, several side effects have been reported, such as myelosuppression, pulmonary toxicity and metabolic disturbances (Refs Reference Cravedi, Ruggenenti and Remuzzi85, Reference Schaffer and Ross86). Treatment of mRCC with mTOR inhibitors was associated with increased blood glucose levels, hypertriglyceridaemia and hypercholesterolaemia (Refs Reference Motzer76, Reference Hudes77). Similarly, the use of mTOR inhibitors after kidney transplantation was linked to elevated cholesterol and triglyceride levels compared with other immunosuppressive regimens and, thus, the subsequent need for lipid-lowering therapy (Ref. Reference Kasiske87). Diabetes mellitus is a frequent complication after solid organ transplantation with an increased risk of graft failure and cardiovascular mortality. Whereas immunosuppressive treatments with glucocorticoids and calcineurin inhibitors are known to result in insulin resistance and impaired insulin secretion, respectively, the role of mTOR inhibitors in the development of diabetes after transplantation is more controversial (Refs Reference Oterdoom88, Reference Oetjen89). Some studies indicate an independent association of mTOR inhibitors with diabetes onset after transplantation, but others did not come to the same conclusion (Refs Reference Johnston90, Reference Teutonico, Schena and Di Paolo91, Reference Veroux92).
Although mTOR inhibitors have been implemented successfully in different clinical settings, they may only be used in the treatment of specific tumour types, and their efficacy might be limited by cellular escape mechanisms such as rapamycin resistance (Ref. Reference Hudes77). Targeting multiple components of the PI3K/AKT pathway might improve antitumour potency and broaden the spectrum of susceptible tumour types. Indeed, based on structural similarities of PI3K and mTOR, newly developed inhibitors aimed at inhibition of both kinases simultaneously are currently under investigation. In addition to dual PI3K–mTOR inhibitors, selective AKT inhibitors are being tested in xenograft mouse models and early Phase I studies. However, inhibitors targeting multiple components might also have more severe side effects with regard to metabolic control. The use of techniques such as antibody-directed drug delivery could allow cell-type-specific targeting of the PI3K/AKT pathway and thus minimise side effects.
Stress response and MAPK signalling in acquired insulin resistance
Although at the core of the problem there is still no satisfying answer to the question of how insulin resistance develops, a widely discussed concept involves Ser/Thr kinases, which can phosphorylate numerous sites in IRS1 and IRS2. Phosphorylation of IRS proteins on Ser/Thr residues can uncouple the activated IR from downstream signal transduction modules (reviewed in Ref. Reference Boura-Halfon and Zick93). This phenomenon potentially depends on three different mechanisms: prevention of docking of IRS to IR, ubiquitylation followed by the proteolytic breakdown of IRS, or prevention of the docking of downstream effectors such as PI3K. Whereas in the former two cases all insulin-induced effects could be abolished, more selective defects might develop in the latter case, dependent on which modules are uncoupled from the activated IR. Multiple negative inputs converge at the level of IRS proteins. Of major importance appears to be the increased secretion of proinflammatory cytokines from adipocytes as observed in obesity. Proinflammatory signalling often involves activation of the inhibitor of κ light polypeptide gene enhancer in B-cells, kinase (IKBKB, IKK)–NF-κB axis, which is now regarded as a critical pathway linking obesity-associated chronic inflammation with insulin resistance. For example, tumour necrosis factor-dependent downregulation of IRS proteins depends on IKK and can be inhibited by aspirin (Ref. Reference Gao94). Indeed, that salicylate can increase insulin sensitivity is an old observation (Ref. Reference Arena, Dugowson and Saudek95). Whereas IKK-knockout mice are embryonic lethal, mice with IKK hemizygosity show lower fasting blood glucose and insulin levels and improved free fatty acid levels relative to littermate controls when placed on a high-fat diet or rendered leptin deficient (Ref. Reference Yuan96). Furthermore, it has been shown that adipocyte-derived factors can act through IKK to induce insulin resistance in skeletal muscle (Ref. Reference Dietze97).
In addition to inflammation, the activation of Ser/Thr kinases with concomitant downregulation of the function of IRS proteins has been observed downstream of various conditions known to be associated with the development of insulin resistance and T2DM, such as hypoxia, endoplasmic reticulum (ER) stress and the generation of reactive oxygen species. Kinases activated under these conditions are also called stress kinases, because their activity positively correlates with the occurrence of imbalances in cellular homeostasis. An increase in circulating cytokines, as observed under systemic low-level inflammation during obesity, can also activate IRS Ser/Thr kinases (Ref. Reference Boura-Halfon and Zick93). Among the kinases targeting IRS are GSK3, S6K, p38 and several isoforms of the PKC family. The PKC family consists of 12 isoforms grouped as atypical PKCs (ζ and λ), conventional PKCs (α, β and γ), novel PKCs (δ, ε, η and θ), and protein kinase Ns (PKN1, PKN2 and PKN3), from which PKCδ, PKCλ/ζ and PKCθ are known to target IRS. One widely discussed case is the activation of JNK downstream of ER stress and the unfolded protein response (Refs Reference Ozcan98, Reference Xu, Spinas and Niessen99). Obese humans and rodents develop ER stress in hepatocytes and adipocytes, leading to JNK-dependent phosphorylation of IRS1 on Ser307 (numbering as in mouse) (Ref. Reference Aguirre100) followed by its ubiquitylation and proteolytic breakdown. Indeed, global or conditional loss of JNK in adipose tissue, skeletal muscle or the brain was found to attenuate diet-induced insulin resistance in mice fed a high-fat diet, supporting the notion of a repressive role for JNK in insulin action (Ref. Reference Sabio and Davis9). Surprisingly, mice in which the target site for JNK in IRS1 (Ser307) was replaced by an alanine were less insulin sensitive, as were mice lacking JNK1 in hepatocytes (Refs Reference Sabio and Davis9, Reference Copps101). The latter two observations indicate that JNK is required for insulin action in hepatocytes, once more underlining the context dependence of insulin signal transduction. The case of JNK exemplifies the dilemma: a significant number of IRS kinases believed to be responsible for the development of insulin resistance are also required for insulin-dependent metabolic control. For example, ERK1/2 are believed to link insulin with cell proliferation, differentiation and the regulation of lipid metabolism, whereas isoforms of PKC may be required for insulin-induced glucose transport (Refs Reference Avruch102, Reference Roth103, Reference Kotzka104, Reference Rydén105, Reference Martin and Parton106, Reference Bandyopadhyay107, Reference Bandyopadhyay108, Reference Sajan109). These intricate interconnections certainly complicate the development of intervention strategies based on MAPKs in the treatment of insulin resistance.
AMPK – an energy sensor targeted in the treatment of metabolic syndrome
When intracellular energy levels are low, cellular metabolism must shift from energy-consuming anabolic processes towards energy-producing catabolic processes. AMPK, a sensor of the availability of intracellular energy, is activated at low energy levels and regulates cellular processes accordingly. This kinase inhibits insulin-stimulated anabolic processes such as de novo lipogenesis and glycogen synthesis. Nevertheless, AMPK activity supports whole-body glucose homeostasis and improves insulin sensitivity by promoting processes such as glucose uptake and energy expenditure. The effects of the widely used antidiabetic drug metformin have been shown to depend largely on activation of AMPK (Ref. Reference Zhou110). Thus, AMPK is currently the only protein kinase targeted in the treatment of metabolic syndrome.
AMPK signalling pathway
AMPK is a heterotrimeric complex consisting of a catalytic α-subunit and two regulatory subunits (β and γ). There are several isoforms of each subunit encoded by individual genes, including PRKAA1 (α1), PRKAA2 (α2), PRKAB1 (β1), PRKAB2 (β2), PRKAG1 (γ1), PRKAG2 (γ2) and PRKAG3 (γ3) (Ref. Reference Carling111). The different isoforms of AMPK subunits are expressed tissue specifically and exert both overlapping and distinct functions (Refs Reference Um112, Reference Mahlapuu113). The AMPK pathway is activated by a variety of physiological stimuli, such as glucose deprivation, hypoxia, oxidative stress and muscle contraction. The common result of these stimuli is a reduction in cellular energy level and an increase in AMP/ATP ratio, which is crucial for AMPK activity. AMPK is also activated by different hormones, including leptin and adiponectin, but the mechanisms by which these hormones activate AMPK are not yet fully elucidated. For full kinase activity, AMPK must be phosphorylated at Thr172 in the catalytic domain of the α-subunit by upstream kinases such as serine/threonine kinase 11 (STK11, LKB1) and calcium/calmodulin-dependent protein kinase kinase β (CAMKKβ). LKB1 is a constitutively active kinase and considered to be the predominant upstream kinase of AMPK, but also phosphorylates 13 other AMPK-related kinases (Ref. Reference Lizcano114). Protein phosphatases (PP2A and PP2C) antagonise upstream kinases and inhibit AMPK activity by dephosphorylation of Thr172. Most importantly, AMPK activity and Thr172 phosphorylation are highly dependent on the intracellular AMP/ATP ratio. AMP and ATP bind to the γ-subunit of AMPK in a competitive manner. When the AMP/ATP ratio is high, binding of AMP to AMPK allosterically activates kinase activity fivefold and induces conformational changes that block the dephosphorylation of Thr172 by PP2A and PP2C, which preserves activation by upstream kinases (reviewed in Refs Reference Carling111, Reference Hardie115, Reference Witczak, Sharoff and Goodyear116). It has been recently proposed that binding of AMP triggers exposure of a myristoyl group at the AMPK β-subunit, which promotes membrane association and primes AMPK for activation by upstream kinases (Ref. Reference Oakhill117). In addition, it was shown that binding of ADP to AMPK protects against dephosphorylation of Thr172, but does not induce allosteric activation of AMPK (Ref. Reference Xiao118). Activated AMPK phosphorylates substrates such as AS160, GYS1, acetyl-CoA carboxylase α (ACACA, ACC) and malonyl-CoA decarboxylase (MLYCD, MCD), thus stimulating glucose uptake, inhibiting glycogen synthesis, inhibiting de novo lipogenesis and enhancing β-oxidation, respectively. AMPK also indirectly inhibits mTORC1, thereby blocking protein synthesis, enhancing respiration and probably improving insulin sensitivity by counteracting mTORC1- and S6K-induced inhibition of IRS1/2 (reviewed in Ref. Reference Witczak, Sharoff and Goodyear116).
Complex role of AMPK isoforms in metabolic control
As mentioned above, the antidiabetic effects of metformin largely depend on AMPK activation. Thus, characterising the role of AMPK isoforms in mammalian physiology is of great importance and a prerequisite for the achievement of more specific and efficient targeting of AMPK compared with metformin. Genetic mutations in elements of the AMPK pathway in humans and their pathophysiological effects in glucose homeostasis are not yet fully characterised. Several polymorphisms in LKB1 and AMPK α2 and γ2 subunits are associated with insulin resistance and T2DM in different subsets of patients (Refs Reference Lopez-Bermejo119, Reference Horikoshi120, Reference Jablonski121). Interestingly, polymorphisms in LKB1, α1-, α2- and β2-subunits of AMPK as well as in AMPK targets myocyte enhancer factor 2A (MEF2A) and MEF2D were found to be associated with reduced response to metformin treatment (Refs Reference Lopez-Bermejo119, Reference Jablonski121). Because metformin is thought to function mainly by activating AMPK, the identified polymorphisms might affect the functions of LKB1, AMPK, MEF2A and MEF2D. However, the physiological and biochemical consequences of the identified polymorphisms remain to be characterised. Apart from that, LKB1 has tumour suppressor functions and its mutation can cause Peutz–Jeghers syndrome, which is characterised by mucocutaneous pigmentation, hamartomatous polyps and increased risk of cancer (Ref. Reference Mehenni122). In addition, mutations in PRKAG2 were shown to cause hypertrophic cardiomyopathy with Wolff–Parkinson–White syndrome owing to a glycogen storage disorder (Refs Reference Murphy123, Reference Kim, Miller and Young124, Reference Arad125).
The complex roles of AMPK isoforms in insulin-sensitive tissues have been studied in transgenic mice. Whereas loss of AMPKα1 did not alter metabolic control in mice, global or tissue-specific loss of individual AMPK isoforms mostly led to impaired glucose homeostasis (Ref. Reference Jorgensen126). PRKAα2-knockout mice were glucose intolerant and insulin resistant and showed impaired glucose uptake on stimulation with the AMPK activator 5-aminoimidazole-4-carboxamide riboside (AICAR) (Refs Reference Viollet10, Reference Jorgensen126). In addition, deletion of Prkaa2 specifically in β-cells resulted in defective glucose-stimulated insulin secretion (Ref. Reference Beall127). Hepatocyte-specific deletion of Prkaa2 in the liver revealed that AMPK inhibits gluconeogenesis and release of glucose in the liver (Ref. Reference Andreelli128). PRKAβ2-knockout mice had reduced maximal and endurance exercise capacities and were more susceptible to diet-induced weight gain and glucose intolerance; PRKAγ3-knockout mice were shown to have impaired AICAR-stimulated glucose uptake (Refs Reference Steinberg129, Reference Barnes130). By contrast, activation of AMPK in the hypothalamus increased food intake, suggesting that inhibition of AMPK in the hypothalamus could protect against obesity-induced insulin resistance (Ref. Reference Andersson131). Indeed, mice lacking AMPKβ1, which is highly expressed in the liver and brain, were protected against diet-induced obesity, insulin resistance and hepatic steatosis, probably because of reduced food intake (Ref. Reference Dzamko132).
These studies show that the effects of AMPK on glucose homeostasis are highly complex as a result of isoform- and tissue-specific functions. Simultaneous modulation of its activity in different tissues can have opposing effects on glucose homeostasis, which could complicate the development of therapeutic approaches directly targeting AMPK. However, isoform- and tissue-specific targeting could also provide a basis for highly specific and effective therapeutic approaches in addition to metformin treatment.
Metformin and AMPK in clinical use
Metformin has been used in the clinic for several decades for the treatment of insulin-resistant and diabetic patients. The drug improves insulin sensitivity, lowers blood glucose and cholesterol levels without risk of acute hypoglycaemia and weight gain, and reduces the risk of diabetes-related complications such as cardiovascular disease (Ref. 133). The notion that metformin elicits its beneficial effects mainly through the activation of AMPK is further underlined by observations of mice with abolished hepatic AMPK activity due to hepatocyte-specific LKB1 deficiency (Ref. Reference Shaw134). Metformin failed to lower blood glucose in these mice, indicating that activation of AMPK through LKB1 in the liver is required (Ref. Reference Shaw134). Nevertheless, several AMPK-independent effects of metformin have been reported (Ref. Reference Zhou110). It was recently shown that metformin can block gluconeogenesis in isolated mouse hepatocytes independently of LKB1 and AMPK (Ref. Reference Foretz135).The mechanism of AMPK activation by metformin is still controversial. One hypothesis is that metformin activates AMPK indirectly by inhibiting complex I of the respiratory chain, which compromises cellular energy production and increases the AMP/ATP ratio (Refs Reference Owen, Doran and Halestrap136, Reference Brunmair137). However, metformin also activates AMPK in an adenine-nucleotide-independent manner (Ref. Reference Hawley138). More recently, it was proposed that metformin activates PKCζ, which phosphorylates LKB1 at Ser428, resulting in nuclear export of LKB1 and activation of AMPK (Ref. Reference Xie139). Metformin may mainly activate AMPK in the liver, muscle and vasculature, because cellular uptake of the drug is dependent on transmembrane transporters such as solute carrier family 22 (organic cation transporter), member 1 (SLC22A1, OCT-1). Whereas OCT-1-deficient mice indeed have a diminished response to metformin, the role of OCT-1 polymorphisms in diabetic patients is controversial (Refs Reference Shu140, Reference Zhou141).
Metformin is used at inconveniently high doses, and its clinical use is restricted in patients with renal or hepatic disease owing to increased risk of lactic acidosis (Ref. 133). Hence, direct activation of AMPK by other means would be an attractive alternative in the treatment of diabetic patients. The AMPK activator A-769662 efficiently lowered blood glucose and triglycerides and transiently reduced body weight gain in mouse models of genetically induced obesity and insulin resistance (Ref. Reference Cool142). In addition, treatment with AICAR was shown to reduce blood glucose levels in diabetic patients. One side effect associated with the activation of AMPK could be increased food intake because of its role in the hypothalamus. However, treatment with metformin reduces body weight in patients by decreasing appetite and food intake (Refs Reference English143, Reference Tsilchorozidou, Batterham and Conway144). The underlying mechanisms remain poorly understood (Refs Reference English143, Reference Tsilchorozidou, Batterham and Conway144). A transient reduction in food intake was reported in obese mice, but not in lean mice treated with A-769662, because this drug may not activate AMPK in the brain (Ref. Reference Cool142). By contrast, increased food intake was observed in mice treated with AICAR (Refs Reference Andersson131, Reference Kim145). A-769662 and AICAR were also shown to have AMPK-independent activity, and possible side effects of long-term treatment have not been assessed (Refs Reference Moreno146, Reference Santidrian147).
Metformin is now also considered for use in cancer therapy. Epidemiological studies have assessed the association between obesity or T2DM and cancer in large populations (Refs Reference Giovannucci148, Reference Calle and Kaaks149). Although intensively investigated, the molecular mechanisms linking cancer with obesity are still not fully elucidated. Chronic hyperinsulinaemia has been suggested to contribute to increased tumour growth, because it may directly activate insulin receptor on (pre-)neoplastic cells or indirectly through promotion of insulin-like growth factor 1 (IGF1) synthesis. Both insulin and IGF1 enhance tumour growth in xenograft models by increasing cell proliferation and inhibiting apoptosis. There is an ongoing debate as to whether the use of insulin analogues in the treatment of obese and diabetic patients could further increase the risk of cancer. Whereas certain insulin analogues do lead to tumour development in rats, their effect in human patients remains controversial (Refs Reference Drejer150, Reference Evans151, Reference Hansen152). In line with the amelioration of obesity and hyperinsulinemia by metformin, observational data showed that its use was associated with a reduced risk of cancer (Refs Reference English143, Reference Jiralerspong153, Reference Evans154). Additionally, it might also inhibit tumor progression by AMPK-mediated inhibition of mTORC1, and possibly also by a Rac GTPase-dependent and AMPK-independent mechanism (Ref. Reference Kalender155). Combined cancer therapy with metformin and drugs targeting the PI3K/AKT pathway might result in the synergistic inhibition of mTORC1. This strategy could also overcome impaired glucose homeostasis resulting from PI3K/AKT pathway inhibition.
Concluding remarks
The insulin signal transduction network and the biochemical properties of its components have been extensively studied. There is increasing knowledge of how PI3K/AKT, MAPK and AMPK signalling controls and how their failure impairs glucose homeostasis. Moreover, studies in transgenic mice have demonstrated that specific modulation of protein kinase signalling can effectively improve glucose homeostasis and protect against obesity, acquired insulin resistance and diabetes. However, very little translation into clinical practice has taken place. Metabolically relevant cellular functions such as glucose transport, lipogenesis, glycogen synthesis and gluconeogenesis are controlled by kinases that do not act exclusively within the insulin signal transduction network. It has emerged that all signal transduction events within a cell are interconnected and that mere description of the network is not sufficient to define mechanisms underlying both context and stimuli specificity. Hence, global modulation of kinase activity by, for example deletion of PTEN and the use of mTOR inhibitors might result in severe side effects such as cancer and impaired metabolic control, respectively. The development of safe kinase-based therapies will probably remain elusive until we understand how cells integrate signalling information to implement context in their respective intracellular signal transduction network. In addition, the development of specific inhibitors is complicated by high structural similarities in the catalytic domains of different protein kinases. Reduced specificity resulting in the inhibition of multiple targets could be beneficial in cancer therapy because it might potentiate toxicity on cancer cells. Inhibitors used for the treatment of metabolic syndrome should, by contrast, be highly specific in order to minimise side effects and allow long-term treatment. Targeting kinases in their inactive state, in which they show higher structural diversity than in their active conformation, or disrupting protein complexes of kinases was suggested for the design of inhibitors with increased specificity (Refs Reference Sunami156, Reference Kaidanovich-Beilin and Eldar-Finkelman157, Reference Pearce, Komander and Alessi158). Tissue-specific targeting by using transmembrane carriers or metabolic activation, as well as the targeting of specific isoforms or effectors further downstream, might provide a route to increased specificity of drugs and minimal side effects.
Acknowledgements
We are grateful to P. King and D. Hynx for a critical reading of the manuscript and to the peer reviewers for their constructive and valuable comments. S.M.S. and O.T. were supported by the Swiss SystemsX.ch initiative LiverX of the Competence Center for Systems Physiology and Metabolic Diseases. O.T. was supported by the Amélie Waring foundation. M.N. was supported through participation in COST Action BM0602 and the Takeda Foundation. The FMI is part of the Novartis Research Foundation.



