


1. Introduction
Model organisms offer us our deepest understanding of many biological phenomena. Scientists are now capitalizing on the knowledge of these model systems to perform comparative studies of the phylogenetic relatives of many model organisms, such as Arabidopsis thaliana (Mitchell-Olds, Reference Mitchell-Olds2001), Caenorhabditis elegans (Harris et al., Reference Harris, Chen, Cunningham, Tello-Ruiz, Antoshechkin, Bastiani, Bieri, Blasiar, Bradnam, Chan, Chen, Chen, Davis, Kenny, Kishore, Lawson, Lee, Muller, Nakamura, Ozersky, Petcherski, Rogers, Sabo, Schwarz, Van Auken, Wang, Durbin, Spieth, Sternberg and Stein2004), Danio rerio (Quigley et al., Reference Quigley, Manuel, Roberts, Nuckels, Herrington, MacDonald and Parichy2005) and Drosophila melanogaster (Singh et al., Reference Singh, Larracuente, Sackton and Clark2009). Such studies promise to unravel the genetic basis of phenotypic evolution and strongly implicate the evolutionary forces responsible for species divergence. Clearly, comparative studies of phenotypic differences between species must be based on a good understanding of the phylogenetic relationships among the taxa involved (Felsenstein, Reference Felsenstein1985).
If the phylogeny is poorly known, the quality of the conclusions from comparative analyses will be poor. Unfortunately, the research traditions of those working on model organisms have not emphasized a phylogenetic framework, leaving us with an inadequate understanding of the relationships of model organisms to some of their close relatives (Al-Shehbaz & Kane, Reference Al-Shehbaz and Kane2002; Kiontke et al., Reference Kiontke, Gavin, Raynes, Roehrig, Piano and Fitch2004; Quigley et al., Reference Quigley, Turner, Nuckels, Manuel, Budi, MacDonald and Parichy2004). The unfortunate result is that our phylogenetic information on clades containing model organisms is often fairly weak, even though these are precisely the clades best suited to comparative studies.
A prime example of this paradox is the genus Drosophila and closely related genera. Over the past 20 years, many studies dealing with parts of the Drosophila phylogeny have been published (for an overview, see van der Linde & Houle, Reference van der Linde and Houle2008), but surprisingly few have adequately addressed the phylogenetic relationships among the subgenera of Drosophila in the context of the various closely related genera. Consequently, even a cursory examination of the literature reveals that many aspects of drosophilid phylogeny are controversial or poorly studied (Ashburner et al., Reference Ashburner, Golic and Hawley2005; Markow & O'Grady, Reference Markow and O'Grady2006).
Grimaldi's (Reference Grimaldi1990) phylogeny based on morphological characters is the most recent comprehensive family wide treatment. An important competing phylogenetic hypothesis is that of Throckmorton (Reference Throckmorton and King1975), which differs from it in many respects. Throckmorton's work was clearly based on many sources of evidence (e.g. Throckmorton, Reference Throckmorton1962, Reference Throckmorton1965, Reference Throckmorton1966); unfortunately, he did not use explicit and reproducible methods. More recently, many phylogenetic hypotheses based on molecular data have been published (see Table 1, van der Linde & Houle, Reference van der Linde and Houle2008, for the most important studies). The best of these have emphasized clades well below the genus level or have been based on small numbers of genes. Figure 1 shows the combination of gene numbers and species numbers in other phylogenetic studies using molecular data. Some aspects of the phylogeny, such as relationships within the melanogaster species subgroup (see Coyne et al., Reference Coyne, Elwyn, Kim and Llopart2004), now seem robustly supported by analysis of large sets of molecular data. At the same time, the results from other studies show that various clades within the phylogeny differ in many key respects.
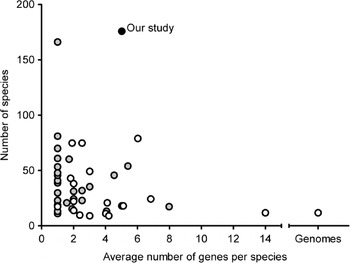
Fig. 1. Overview of average number of genes per species and number of species included in previous studies compared with those in our study. Black dot: our study; grey dots: studies limited to a single genus or Drosophila subgenus or major clade within the subgenus Drosophila (e.g. immigrans-tripunctata radiation, virilis-repleta radiation and Hawaiian Drosophila); white dots: studies covering at least two genera and/or Drosophila subgenera and/or major clades of the subgenus Drosophila.
Table 1. ML estimates of parameter values for each data partition under the GTR+Γ model
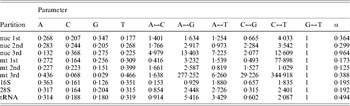
Parameter values include the base frequencies and the instantaneous substitution rates between nucleotides. The G↔T is set to 1·0 by default. α is the value of the shape parameter for the Γ distribution describing among-site rate variation. Data partitions include the codon positions for protein coding regions of each genome (nuclear, ‘nuc’; mitochondrial, ‘mt’). Values were calculated by RAxML as described in the text. Particular values of note include the very strong AT bias in mt 3rd positions in contrast to the GC bias in nuc 3rd positions, much less even substitution rates at 3rd positions of both genomes, and the weak transition/transversion ratio in 16S.
The underlying source of this lack of consensus is that the available data are fragmentary. Taxon sampling of this very large group has been haphazard, and a few studies sequence more than a small number of genes. The result is that the sequence available for a pair of closely related species is likely to come from different genes, making meaningful phylogenetic analyses difficult. We refer to this situation as a lack of overlap.
In this situation, phylogenetic hypotheses can be generated through a supertree analysis of published results (see, e.g. Bininda-Emonds et al., Reference Bininda-Emonds, Gittleman and Steel2002; Bininda-Emonds, Reference Bininda-Emonds2004). Our supertree analysis and review covering Drosophila and its closely related genera (van der Linde & Houle, Reference van der Linde and Houle2008) resulted in a generally well-resolved tree and clearly showed that the genus Drosophila as currently defined is paraphyletic with respect to various genera (e.g. Scaptomyza, Hirtodrosophila, Samoaia and Zaprionus) placed within it (Fig. 2). Supertree methods have been criticized on various grounds, including that they give too much weight to weakly supported or erroneous nodes, difficulty in dealing with biased data, failure to use the original data, inapplicability of model-based methods of analysis and failure to use all the data efficiently (Kluge, Reference Kluge1989; Gatesy et al., Reference Gatesy, Baker and Hayashi2004; but see also Bininda-Emonds et al., Reference Bininda-Emonds, Jones, Price, Grenyer, Cardillo, Habib, Purvis and Gittleman2003; Bininda-Emonds, Reference Bininda-Emonds2004). Some of these issues are apparent in our own supertree analysis (van der Linde & Houle, Reference van der Linde and Houle2008).

Fig. 2. The phylogenetic relationships as understood on the basis of the available literature before the study reported here.
Here, we report the results of a supermatrix analysis in which we used publicly available sequence data, plus a limited amount of new sequence chosen to increase overlap. Our data stand out in the number of species included (180; Fig. 1) and in the variety of taxa included. We note that, although ours is the most comprehensive study to date, overlap of the sequence data for many species remains limited. The focus of the study is to resolve the basal nodes within the genus Drosophila sensu lato. The major issue is the topology of the three main clades of the subgenus Drosophila and the various genera that are positioned among them (e.g. Hirtodrosophila, Zaprionus and Scaptomyza). In addition, the order of the two genera placed sister to the genus D. sensu lato (Scaptodrosophila and Chymomyza) is still insufficiently resolved (Tarrio et al., Reference Tarrio, Rodriguez-Trelles and Ayala2001). The phylogenetic relationships between the various species groups in both the immigrans-tripunctata clade and the virilis-repleta clade are not yet fully resolved. Finally, the monophyletic nature of many groups is questioned.
2. Materials and methods
(i) Species and data
We compiled data for 541 species: 535 species in family Drosophilidae and six outgroup species in the families Tephritidae (one species), Ephydridae (four) and Lauxaniidae (one). We screened loci in GenBank for which multiple drosophilids had been sequenced and selected 13 to maximize taxonomic overlap among gene sequences. These include nine nuclear loci (Adh, AmyRel, per, Ddc, Sod, yp1, 28Sd1, 28Sd2 and 28Sd8) and four mitochondrial loci (COI, COII, COIII and 16S). Most sequences were obtained from GenBank, supplemented with new sequences for COI, COII, COIII, 28Sd1 and 28Sd8. Only protein-coding regions of the non-ribosomal genes were used. The aligned sequences for AmyRel were kindly provided by J.-L. Da Lage. After generating the full set of data, we selected species on the basis of number of genes available, number of base pairs and our need for a distance matrix covering a large number of species for which we have wing-shape data. On the basis of these criteria, we selected 180 species (see Supplementary Table 1). The total number of base pairs per species ranged from 339 to 13 539 bp (mean: 4333 bp); only seven taxa had fewer than 1000 bp. Accession codes can be found in Supplementary Table 1; new sequences are available from GenBank under accession codes GU597372 to GU597535. Collection locations of the 39 species for which we collected new sequences can be found in Supplementary Table 2.
(ii) Alignment
Nucleotide sequences were aligned with Clustal X (Thompson et al., Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997) and manually inspected in MacClade (Maddison & Maddison, Reference Maddison and Maddison2005) for resolution of regions of ambiguity or disagreement and consolidated indels. Alignment of all protein-coding regions was trivial because amino acid indels were rare and readily interpreted. Sequences for the genes were concatenated for each taxon. The alignment and trees [maximum parsimony (MP), partitioned maximum-likelihood (ML) and Bayesian] are available from TreeBase under accession code SN4940.
(iii) Phylogenetic analysis
Heterogeneity of nucleotide composition among informative sites was estimated with PAUP* version 4.0b10 (Swofford, Reference Swofford2002). Only two of the 13 genes, AmyRel and Adh, showed heterogeneity. Phylogenetic analyses were conducted under MP, neighbour-joining (NJ), ML and Bayesian approaches. Because of the number of species without overlapping data, we did not test for congruence using, for example, a partition-homogeneity test (Farris et al., Reference Farris, Kallersjo, Kluge and Bult1994, Reference Farris, Kallersjo, Kluge and Bult1995). All MP analyses were conducted with PAUP* version 4.0b10 (Swofford, Reference Swofford2002) with heuristic searches with tree bisection-reconnection (TBR) branch swapping and 20 random-addition starting trees; the first 2000 trees found were retained. All substitutions were weighted equally; gaps were treated as missing data. To determine whether changes in base composition in AmyRel and Adh might mislead phylogenetic reconstruction, we analysed these genes separately using NJ with LogDet distances in PAUP* and compared the results to the ML and Bayesian trees for well-supported conflicts. LogDet is less sensitive to base-composition heterogeneity (Swofford et al., Reference Swofford, Olsen, Waddell, Hillis, Hillis, Moriz and Mable1996). Although the LogDet NJ tree and the ML/Bayesian trees differed, the differences were not well supported, and the ML gene tree was not appreciably more similar to the concatenated tree than was the LogDet NJ tree, suggesting that convergence on nucleotide frequencies was not misleading the combined-data analyses.
We conducted ML analyses in several ways to account for the complexity of the data. First, a series of analyses on unpartitioned data was conducted with PAUP*. ML parameter values were estimated under a nested array of substitution models for a random MP tree as implemented in Modeltest 3.04 (Posada & Crandall, Reference Posada and Crandall1998); likelihood-ratio tests (Yang et al., Reference Yang, Goldman and Friday1995) and the Akaike Information Criterion (Akaike, Reference Akaike1973) were used to identify the simplest models of sequence evolution that adequately fit the data and phylogeny. The most complex model was selected by both criteria: GTR+I+Γ. Parameters were fixed for the values estimated on the initial MP tree. Heuristic searches were then conducted with two alternatives for generating starting trees. In the first search, the first 20 trees were saved from each of 10 random-addition replicates from MP analyses (200 maxtrees total), and these 200 trees became the starting trees for the ML search, providing some initial sampling of tree space. MP trees were used rather than a single NJ tree because non-overlapping data among sets of taxa resulted in undefined distances and anomalous placement of some taxa in the NJ tree. Parsimony appeared less affected by non-overlapping data, in that recovered trees were consistent with published studies. In addition, five parallel random-sequence-addition runs were conducted, each of which required approximately 200 h to build the starting trees. The search starting with the MP trees yielded a more likely tree (ln L=−178603 as opposed to −178606), and that tree is reported here.
Our data include protein-coding and RNA genes from mitochondrial and nuclear genomes, so a single model may be insufficient to account for the data. We therefore conducted partitioned ML analyses using RAxML (Stamatakis et al., Reference Stamatakis, Hoover and Rougemont2008) using the Cipres Portal 1.15 to access the San Diego Supercomputer Center. Conditions followed the default settings, including a GTR+Γ model, and no per-gene branch length optimization (branch lengths were proportional across partitions). We partitioned the data by codon position separately for the three nuclear codon positions and the three mitochondrial codon positions, as well as by genome for the ribosomal genes and for the tRNAs, producing nine partitions (1st, 2nd and 3rd nuclear; 1st, 2nd and 3rd mitochondrial; mt rRNA, nu rRNA and tRNA). We repeated the analyses unpartitioned for comparison with the PAUP* results to distinguish the effects of partitioning from software-specific tree-search strategies. Parameter values as estimated by RAxML for each partition are reported in Table 1.
Non-parametric bootstrapping (Felsenstein, Reference Felsenstein1985) was performed under ML with two approaches, one using the genetic algorithm approach in GARLI version 0.951 (Zwickl, Reference Zwickl2006) with the data unpartitioned and the other using RAxML to allow partitioned data. Garli analyses used 200 replicates and the GTR+I+Γ model with parameters estimated by GARLI. The search was conducted with a random starting tree and an automatic run termination after a minimum of 5000 generations that did not improve topology, a ln L improvement of less than 0·02 due to topological changes, a 0·05 score improvement threshold, and default genetic algorithm settings. The second bootstrapping approach used RAxML with both unpartitioned and partitioned data sets. Bootstrapping was run for 250 replicates (RAxML selected 150 as sufficient), representing the combined output of two independent runs (100 and 150 replicates) with different starting seeds, with default settings. Partitioning was the same as in the ML searches. MP bootstrapping was conducted with 500 replicates, and 50 trees were saved in each random-addition replicate.
Bayesian analyses used the mpi (multiple processors) version of MrBayes 3.2 (Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003) distributed over eight processors. We partitioned the data as with RAxML. Two independent analyses of four heated chains each were run for 80 million generations. Parameters were estimated for each partition separately (‘unlinked’); trees and parameters were recorded every 1000 generations. Convergence was estimated by means of cumulative and sliding plots from “Are We There Yet?” (AWTY) (Wilgenbusch et al., Reference Wilgenbusch, Warren and Swofford2004) as well as by examination of likelihood plots and posterior probabilities of individual clades for subsets of the runs. The chains converged slowly on the basis of the AWTY and likelihood plot diagnostics, possibly because large blocks of data were missing and overlap among several taxa was limited, yielding a burn-in of 50%. Split-frequency standard deviations never indicated full convergence, plateauing around 0·1 by 20 million generations. We calculated a majority-rule consensus tree of the post-burn-in trees from both runs to summarize posterior probabilities.
3. Results and discussion
At the level of the previously recognized genera, the results of the ML and Bayesian analyses were strongly concordant with each other (Fig. 3). The main differences between the analyses are in the placement of the immigrans-tripuncata, Zaprionus and Hirtodrosophila/Mycodrosophila clades and among the poorly supported nodes near the root of immigrans-tripuncata clade. The concordance between the ML and Bayesian analyses within genera differed dramatically for different genera. The MP results were also largely concordant, lacking any significant conflict (MP bs >80%); the strict-consensus MP tree and the ML tree had 140 nodes in common. All but three differences (all in subgenus Drosophila, discussed individually below) were confined to regions poorly supported in all analyses and with <42% MP bootstrap support. MP bootstrap values were strongly correlated with the other support values and were nearly always the lowest of the four values. Because MP bootstrap values do not provide a strong independent signal, only the model-based values are reported on Fig. 3.

Fig. 3. Partitioned ML phylogram of the Drosophilidae. Taxon names along branches are the traditional classification, whereas names within the coloured boxes are the proposed genus names for species currently assigned to Drosophila. Branches coloured by current genus; Drosophila appears in black. Support values above branches are unpartitioned ML bootstrap (bs; PAUP), partitioned ML bs (RAxML) and Bayesian posterior probabilities (pp), respectively, expressed as percentages. Only bs >70 or pp >85 are shown. Symbols: ‘*’ indicates 100; ‘—’ indicates the node is below 50% in the bootstrap majority tree and not present in the Bayesian majority rule tree.
(i) Steganinae and Drosophilinae
Traditionally, the family Drosophilidae is split into two subfamilies, the Drosophilinae and the Steganinae (Hendel, Reference Hendel1917; Duda, Reference Duda1924; Throckmorton, Reference Throckmorton1962, Reference Throckmorton1965, Reference Throckmorton and King1975; Okada, Reference Okada1989; Grimaldi, Reference Grimaldi1990; Sidorenko, Reference Sidorenko2002). Our small sample of subfamily Steganinae (four taxa) suggests that it may be paraphyletic with respect to the subfamily Drosophilinae. Although the Steganinae are monophyletic in the partitioned ML tree (Fig. 3 a), the single species of the genus Leucophenga is the sister taxon to the Drosophilinae in the Bayesian and the unpartitioned ML analyses. The node supporting paraphyly was poorly supported [ML unpartitioned bootstrap (MLu bs): 42; Bayesian posterior probability (pp), reported as a percentage: 81] relative to the nodes for the family Drosophilidae (MLu bs: 95; pp: 100) and subfamily Drosophilinae (MLu bs: 58; pp: 100).
Previous analyses of these subfamilies suggest that no single character distinguishes the two subfamilies (see Ashburner et al., Reference Ashburner, Golic and Hawley2005, for discussion). The only molecular study that has included species of both subfamilies (Remsen & O'Grady, Reference Remsen and O'Grady2002) confirmed this basal subdivision, although one of the unrooted molecular trees (16S) also suggests that this subfamily is paraphyletic. Our results stress the need for additional sampling of species from both subfamilies before any conclusions regarding the basal nodes within the family Drosophilidae can be drawn.
(ii) Scaptodrosophila and Chymomyza
Our analyses are equivocal with respect to the positions of Chymomyza and Scaptodrosophila, and the two alternative arrangements are both poorly supported. Chymomyza appears monophyletic [two species, MLu bs, ML partitioned bootstrap (MLp bs) and pp support all 100], whereas Scaptodrosophila is paraphyletic with respect to two moderately to well-supported clades (Scaptodrosophila deflexa plus Scaptodrosophila lebanonensis group, 99–100 for all analyses; Scaptodrosophila dorsocentralis/latifasciaformis only moderate). Examination of the underlying data reveals a very limited overlap in sequences between species of the two Scaptodrosophila clades, which could explain the lack of support for monophyly of the genus.
Scaptodrosophila and Chymomyza appear to be the closest relatives to Drosophila for which sequences are available. Most morphological (Okada, Reference Okada1963; Hu & Toda, Reference Hu and Toda2001) and molecular (DeSalle, Reference DeSalle1992 a; Kwiatowski et al., Reference Kwiatowski, Skarecky, Bailey and Ayala1994, Reference Kwiatowski, Krawczyk, Jaworski, Skarecky and Ayala1997) studies do not contradict Tarrio et al. (Reference Tarrio, Rodriguez-Trelles and Ayala2001), who suggested that Scaptodrosophila diverged from other drosophilids before Chymomyza did, on the basis of almost 5000 bp of sequence from five nuclear genes. Remsen & O'Grady (Reference Remsen and O'Grady2002) found that Scaptodrosophila and Chymomyza formed a sister clade to the Sophophora but with low support. Other studies have been unable to resolve this node (Throckmorton, Reference Throckmorton and King1975; Grimaldi, Reference Grimaldi1990; Remsen & DeSalle, Reference Remsen and DeSalle1998; Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). Our results do not provide clear support for any of the topologies and raise questions about the monophyly of the genus Scaptodrosophila. These issues can only be resolved with further data.
(iii) Genus Drosophila and included genera
The genus Drosophila, together with the included genera, forms a moderately supported clade (MLu bs: 58; MLp bs: 79; pp: 100), and most species are placed in two major clades. One is the monophyletic subgenus Sophophora (MLu bs: 58; MLp bs: 79; pp: 100; Fig. 3 a). The remaining genera and subgenera form a separate well-supported clade (MLu bs: 93; MLp bs: 95; pp: 100; Fig. 3 b), within which most species are distributed over two major clades. The first major clade contains the Hawaiian drosophilids – Hawaiian Drosophila clade and Scaptomyza – and the virilis-repleta radiation and is well supported (MLu bs: 88; MLp bs: 96; pp: 87). The two Hawaiian clades are each monophyletic and sister to each other (98–100 for all analyses). The sister taxon to the Hawaiian drosophilids is the virilis-repleta radiation, which is monophyletic in all analyses with strong support (MLu bs: 94; MLp bs: 100; pp: 87).
The second major clade contains three groups (Fig. 3 b), the immigrans-tripunctata radiation, Zaprionus and Hirtodrosophila/Mycodrosophila. Together with some of the smaller genera, they form a weakly supported clade (MLu bs: 51; MLp bs: 68; pp: 87). Unfortunately, the analyses failed to resolve the topology among the three basal lineages consistently. The immigrans-tripunctata radiation was monophyletic in all analyses (MLu bs: 81; MLp bs: 100; pp: 100), as was the genus Zaprionus (MLu bs: 89; MLp bs: 90; pp: 98). The Hirtodrosophila and Mycodrosophila species, except H. duncani, form a clade (MLu bs: 89; MLp bs: 94; pp: 99). The genus Liodrosophila was placed in the sister clade of the genus Zaprionus in all analyses, although with weak support.
The subgenus Dorsilopha and the genus Dettopsomyia were placed basal to these two major clades. In the unpartitioned ML analysis, the two genera form a clade with weak bootstrap support (61%), whereas in the Bayesian analysis, they are successive outgroups to the major lineage, but the pp for the relevant node is only 62%.
The largest Drosophila phylogeny to date with respect to number of genes used is based on the genomes of the 12 sequenced species (Drosophila 12 Genomes Consortium, 2007). The topology of the 12-genome study is identical with our topology for the same species, underlining the robustness of our analysis. The subgenus Sophophora is the sister clade of the remaining subgenera, as well as of the regularly included genera (Beverley & Wilson, Reference Beverley and Wilson1984; DeSalle, Reference DeSalle1992 b; Wojtas et al., Reference Wojtas, Vonkalm, Weaver and Sullivan1992; Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Thomas & Hunt, Reference Thomas and Hunt1993; Kwiatowski et al., Reference Kwiatowski, Skarecky, Bailey and Ayala1994, Reference Kwiatowski, Krawczyk, Jaworski, Skarecky and Ayala1997; Russo et al., Reference Russo, Takezaki and Nei1995; Remsen & DeSalle, Reference Remsen and DeSalle1998; Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999; Tarrio et al., Reference Tarrio, Rodriguez-Trelles and Ayala2001; Remsen & O'Grady, Reference Remsen and O'Grady2002; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). The position of the major clades in the subgenus Drosophila combined with the included genera is not recovered consistently, but our results support the grouping of the Hawaiian Drosophila clade with Scaptomyza (Throckmorton, Reference Throckmorton and King1975; DeSalle, Reference DeSalle1992 a; Thomas & Hunt, Reference Thomas and Hunt1993; Kambysellis et al., Reference Kambysellis, Ho, Craddock, Piano, Parisi and Cohen1995; Russo et al., Reference Russo, Takezaki and Nei1995; Remsen & DeSalle, Reference Remsen and DeSalle1998; Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999, Reference Tatarenkov, Zurovcova and Ayala2001; Davis, Reference Davis2000; Davis et al., Reference Davis, Kurihara, Yoshino and Yamamoto2000 b; Remsen & O'Grady, Reference Remsen and O'Grady2002; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; O'Grady & DeSalle, Reference O'Grady and DeSalle2008), which together form the sister clade of the virilis-repleta radiation (Kambysellis et al., Reference Kambysellis, Ho, Craddock, Piano, Parisi and Cohen1995; Russo et al., Reference Russo, Takezaki and Nei1995; Remsen & DeSalle, Reference Remsen and DeSalle1998; Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999, Reference Tatarenkov, Zurovcova and Ayala2001; Gailey et al., Reference Gailey, Ho, Ohshima, Liu, Eyassu, Washington, Yamamoto and Davis2000; Tarrio et al., Reference Tarrio, Rodriguez-Trelles and Ayala2001; Tatarenkov & Ayala, Reference Tatarenkov and Ayala2001; Remsen & O'Grady, Reference Remsen and O'Grady2002; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). Most studies recover the remaining three major clades – Zaprionus, the immigrans-tripunctata radiation and Hirtodrosophila/Mycodrosophila – as sister taxa to each other, but no consensus has emerged about their branching order (Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999; Davis et al., Reference Davis, Kurihara and Yamamoto2000 a; Gailey et al., Reference Gailey, Ho, Ohshima, Liu, Eyassu, Washington, Yamamoto and Davis2000; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007).
(iv) Subgenus Sophophora
Our analyses recover the previously identified Neotropical and Old World clades within the subgenus Sophophora. The Neotropical clade contains the willistoni and saltans species groups (all support values: 100), whereas the ‘Old World’ clade contains the obscura, ananassae, montium and melanogaster species groups (MLu bs: 71; MLp bs: 94; pp: 100). The obscura and ananassae species groups form sequential sister groups to the montium and melanogaster species groups (cf. Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). Each of the six species groups was monophyletic (MLbs: 96–100; pp: 100), and the topology was well supported in both the ML and Bayesian analyses.
The subgenus Sophophora has been traditionally split into eight species groups, of which the four largest – melanogaster, obscura, saltans and willistoni – are generally included in phylogenetic studies (Pitnick et al., Reference Pitnick, Markow and Spicer1999; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999; Bächli, 1999–Reference Bächli2009; O'Grady & Kidwell, Reference O'Grady and Kidwell2002; Remsen & O'Grady, Reference Remsen and O'Grady2002; Ashburner et al., Reference Ashburner, Golic and Hawley2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). Recently, Da Lage et al. (Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007) proposed to elevate the montium and ananassae species subgroups to the level of species groups, bringing the total number to 10. The 10 groups are distributed among the two major clades, one containing the melanogaster, montium, ananassae and obscura species groups and the other containing the willistoni and saltans species groups (Pélandakis et al., Reference Pélandakis, Higgins and Solignac1991; Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Russo et al., Reference Russo, Takezaki and Nei1995; Kwiatowski & Ayala, Reference Kwiatowski and Ayala1999; Pitnick et al., Reference Pitnick, Markow and Spicer1999; O'Grady & Kidwell, Reference O'Grady and Kidwell2002; Remsen & O'Grady, Reference Remsen and O'Grady2002; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007).
Our study confirms the generally accepted topology of the subgenus Sophophora, as well as the validity of the proposal by Da Lage et al. (Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007) to split the melanogaster species group into three: ananassae, montium and melanogaster. Among these three species groups, the montium and melanogaster groups are sister to each other (MLu bs: 83; MLp bs: 97; pp: 100). This topology has been observed in many studies (cf. Hsu, Reference Hsu1949; Inomata et al., Reference Inomata, Tachida and Yamazaki1997; Goto & Kimura, Reference Goto and Kimura2001; O'Grady & Kidwell, Reference O'Grady and Kidwell2002; Kastanis et al., Reference Kastanis, Eliopoulos, Goulielmos, Tsakas and Loukas2003; Lewis et al., Reference Lewis, Beckenbach and Mooers2005 b; Kopp, Reference Kopp2006; Prud'homme et al., Reference Prud'homme, Gompel, Rokas, Kassner, Williams, Yeh, True and Carroll2006; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007). In this context, the placement of the fima species group as the sister clade of the ananassae species subgroup (Pélandakis & Solignac, Reference Pélandakis and Solignac1993) provides an additional argument for accepting the proposed split of the melanogaster species group.
The subgenus Sophophora as currently defined is already recognized as paraphyletic; the genus Lordiphosa (not included in our study) is the sister clade of the willistoni-saltans clade (Katoh et al., Reference Katoh, Tamura and Aotsuka2000; Hu & Toda, Reference Hu and Toda2001). In our study, the only member of the duncani species group, H. duncani (Wheeler, Reference Wheeler1949), was also placed in this subgenus as the sister to the willistoni-saltans clade.
(v) Virilis-repleta radiation
Most studies suggest division of the virilis-repleta radiation into repleta and virilis clades (Pitnick et al., Reference Pitnick, Markow and Spicer1999; Katoh et al., Reference Katoh, Tamura and Aotsuka2000; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006). In our analyses, the virilis species group was placed sister to the repleta clade with high support in the Bayesian analysis (pp=100) but not in the ML analyses (MLu bs: 48; MLp bs: 67). The repleta clade, which includes one of the two species of the subgenus Siphlodora (Drosophila flexa), was reasonably well supported (MLbs: 82; MLp bs: 88; pp: 79), but the topology of the species groups within the clade differed slightly in the MP, ML and Bayesian analyses, primarily in the position of the dreyfusi species group (Drosophila camargoi). Generally, the support for the nodes dealing with the placement of the various species groups is low. Monophyly for most of the species groups is strongly supported, except for the robusta species group, which is paraphyletic, in line with a previous study including this group (Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006).
The topology within the virilis-repleta radiation is poorly resolved, although some consensus on aspects of the tree has emerged. The robusta, melanica, angor and quadrisetata species groups generally form a clade (MLu bs: 94; MLp bs: 100; pp: 90) (Watabe & Peng, Reference Watabe and Peng1991; Pitnick et al., Reference Pitnick, Markow and Spicer1999; Remsen & O'Grady, Reference Remsen and O'Grady2002; Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006), whereas the repleta clade consists of the repleta, mesophragmatica, bromeliae, dreyfusi, annulimana, flavopilosa and canalinea species groups (Pitnick et al., Reference Pitnick, Markow and Spicer1999; Tatarenkov & Ayala, Reference Tatarenkov and Ayala2001; Remsen & O'Grady, Reference Remsen and O'Grady2002; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007) with moderate support (MLu bs: 82; MLp bs: 88; pp: 79). The studies differ in the position of the virilis species group; Pitnick et al. (Reference Pitnick, Markow and Spicer1999) and Wang et al. (Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006) place it sister to the robusta-melanica clade, whereas others place it sister to the repleta clade (Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Tatarenkov & Ayala, Reference Tatarenkov and Ayala2001; Remsen & O'Grady, Reference Remsen and O'Grady2002). Our results favour its placement sister to the repleta clade (MLu bs: 48; MLp bs: 67; pp: 100), but further study is clearly needed to resolve this issue. The nannoptera species group is generally placed within the repleta clade (Pitnick et al., Reference Pitnick, Markow and Spicer1999; Tatarenkov & Ayala, Reference Tatarenkov and Ayala2001; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006), whereas Robe et al. (Reference Robe, Valente, Budnik and Loreto2005) place it outside the whole genus on the basis of a Bayesian analysis of the amd gene, while Remsen & O'Grady (Reference Remsen and O'Grady2002) place it sister to the melanica-robusta clade.
D. flexa belongs to the small subgenus Siphlodora, which has only been included in a single analysis before ours (Remsen & O'Grady, Reference Remsen and O'Grady2002), where it was placed as the sister species of the nannoptera species group. Our results show it to be a basal member of the repleta lineage, which includes the nannoptera species group.
Parsimony differs from the model-based results, showing moderate conflict in two respects. MP places Drosophila hydei sister to Drosophila hamatofila (MP bs: 63%) and Drosophila buzzatii sister to the Drosophila mulleri/mojaviensis clade (MP bs: 54%).
(vi) Immigrans-tripunctata radiation
Our results place the immigrans clade (bs and pp: 100) as the sister group to the tripunctata/funebris groups (MLu bs: 62; MLp bs: 89; pp: 100), but relationships within the tripunctata/funebris clade are unstable, and many nodes are poorly supported. In agreement with previous studies, the tripunctata and guarani clades are not monophyletic (Frota-Pessoa, Reference Frota-Pessoa1954; Throckmorton, Reference Throckmorton and King1975; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Yotoko et al., Reference Yotoko, Medeiros, Solferini and Klaczko2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; Hatadani et al., Reference Hatadani, McInerney, de Medeiros, Martins Junqueira, de Azeredo-Espin and Klaczko2009). The broad coverage of species in this study also suggests that the testacea and funebris species groups are not monophyletic. Drosophila bizonata (bizonata species group) is placed within the testacea species group, whereas the two species of the funebris species group are placed at different locations in the tree. Additional work is needed in this group.
Previous studies are generally consistent regarding the major splits in the immigrans-tripunctata radiation. All but two (Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; Katoh et al., Reference Katoh, Nakaya, Tamura and Aotsuka2007) have concluded that the immigrans-tripunctata group is monophyletic. Most authors place the immigrans species group sister to the remaining members of the immigrans-tripunctata radiation (Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Remsen & O'Grady, Reference Remsen and O'Grady2002; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Perlman et al., Reference Perlman, Spicer, Shoemaker and Jaenike2003; Yotoko et al., Reference Yotoko, Medeiros, Solferini and Klaczko2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005), but mirroring our results, previous studies are equivocal about relationships within the tripunctata/funebris clade (Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Remsen & O'Grady, Reference Remsen and O'Grady2002; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Yotoko et al., Reference Yotoko, Medeiros, Solferini and Klaczko2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; Hatadani et al., Reference Hatadani, McInerney, de Medeiros, Martins Junqueira, de Azeredo-Espin and Klaczko2009). Additional studies with a better coverage of species will be required before the relationships between the various groups can be resolved.
(vii) Drosophila repletoides
Yassin and co-workers (Yassin, Reference Yassin2007; Yassin et al., Reference Yassin, Araripe, Capy, Da Lage, Klaczko, Maisonhaute, Ogereau and David2008, Reference Yassin, Da Lage, David, Kondo, Madi-Ravazzi, Prigent and Toda2010) have shown that the tumiditarsus species group, which contains D. repletoides, is positioned close to the genus Zaprionus and that several species of the Zaprionus subgenus Anaprionus actually belong to the tumiditarsus species group. Our only representative of the subgenus Anaprionus has not been affected by this taxonomic change, and is closely related to the remaining species of the genus (cf. Yassin et al., Reference Yassin, Da Lage, David, Kondo, Madi-Ravazzi, Prigent and Toda2010; Amir Yassin, personal communication). Our model-based analyses indicate that D. repletoides is most probably in a clade with the genera Zaprionus and Liodrosophila (MLu bs: 71; MLp bs: 52; pp: 83), together sister to the immigrans-tripunctata radiation. In contrast, the MP analysis weakly places it sister to the Hawaiian Drosophila/Scaptomyza/Dettopsomyia clade, with no intervening node with greater than 27% MP bs. The ML and Bayesian results are more decisive; several intervening clades are well supported (e.g. the virilis-repleta radiation/Hawaiian Drosophila/Scaptomyza clade).
(viii) Paraphyly of taxa
Our results confirmed that several species groups, e.g. tripunctata, guarani, testacea, robusta and funebris, as well as genera, e.g. Drosophila and Hirtodrosophila, were paraphyletic or even polyphyletic. Our results indicated that Hirtodrosophila is not monophyletic, as suggested by Bächli et al. (Reference Bächli, Vilela, Escher and Saura2004), whereas the monophyly of Scaptodrosophila is unclear. For Scaptodrosophila, the paraphyly might just be an artefact of the underlying data (see above), in that the overlap in sequences between the two clades is relatively small. The situation for Hirtodrosophila is clearly different. In our analysis, the overlap in the sequences between H. duncani and Hirtodrosophila thoracis spans four different genes. H. duncani is placed within the subgenus Sophophora sister to the willistoni and saltans species groups, whereas H. thoracis is grouped with the other Hirtodrosophila species, nested within Mycodrosophila. Previous work has suggested this placement of H. duncani. It was placed in its own unique species group on the basis of its unique male genitalia (Wheeler, Reference Wheeler1949). Nater (Reference Nater1950, Reference Nater1953) concluded that the male genitalia are most similar to those of the obscura subgroup. Burla (Reference Burla1956) noted that, among Hirtodrosophila, this species is closest to Drosophila in several internal and external morphological characteristics, whereas Throckmorton (Reference Throckmorton1962) placed it close to or in Sophophora. Finally, Grimaldi (Reference Grimaldi1990) concluded that apart from ‘the presence of the long sensilla trichodea on the first flagellomere … Drosophila duncani has no other Hirtodrosophila features’.
Several species groups were paraphyletic or polyphyletic as well. The most striking is the tripunctata species group, whose members were scattered within the immigrans-tripunctata radiation. This result is in agreement with previous studies (Frota-Pessoa, Reference Frota-Pessoa1954; Throckmorton, Reference Throckmorton and King1975; Carrasco et al., Reference Carrasco, Prado and Godoy-Herrera2003; Yotoko et al., Reference Yotoko, Medeiros, Solferini and Klaczko2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; Hatadani et al., Reference Hatadani, McInerney, de Medeiros, Martins Junqueira, de Azeredo-Espin and Klaczko2009). Additional studies are needed on this group.
The guarani species group is paraphyletic with two major species subgroups – guarani (including Drosophila ornatifrons and guaru) and guaramun (Drosophila maculifrons) – positioned in different locations of the immigrans-tripunctata radiation. The support for splitting this group was especially strong in the Bayesian analysis (two intervening nodes with pp >95), but weak in the ML analyses (largest intervening MLp bs=50%). This result agrees with previous studies (Kastritsis, Reference Kastritsis1969; Clayton & Wheeler, Reference Clayton, Wheeler and King1975; Throckmorton, Reference Throckmorton and King1975; Yotoko et al., Reference Yotoko, Medeiros, Solferini and Klaczko2003; Robe et al., Reference Robe, Valente, Budnik and Loreto2005; Hatadani et al., Reference Hatadani, McInerney, de Medeiros, Martins Junqueira, de Azeredo-Espin and Klaczko2009); treating both subgroups as species groups seems to be fully justified.
A third species group suspected to be polyphyletic is the robusta group (Wang et al., Reference Wang, Park, Watabe, Gao, Xiangyu, Aotsuka, Chen and Zhang2006), and this conclusion was corroborated in our analysis as well. An unexpected find in the immigrans-tripunctata radiation was the placement of the single included species of the bizonata species group (D. bizonata) within the testacea species group. Before our study, no analysis has included members of the bizonata group together with multiple species of the testacea group. The support in both the ML (MLu bs: 90; MLp bs: 98) and the Bayesian analyses is strong (100). Finally, the funebris species group was not monophyletic. The two subgroups – funebris and macrospina – were positioned at different locations within the phylogeny, although the ML and Bayesian analyses differed in the exact position. Support was strong for a closer relationship of Drosophila funebris with Drosophila pinicola than with Drosophila macrospina (MLu bs: 85; MLp bs: 100; pp: 100).
The paraphyletic nature of the genus Drosophila has been reported before by various authors (Throckmorton, Reference Throckmorton1962, Reference Throckmorton1965, Reference Throckmorton and King1975; Beverley & Wilson, Reference Beverley and Wilson1984; Grimaldi, Reference Grimaldi1990; DeSalle, Reference DeSalle1992 a, Reference DeSalleb; Pélandakis & Solignac, Reference Pélandakis and Solignac1993; Thomas & Hunt, Reference Thomas and Hunt1993; Kwiatowski et al., Reference Kwiatowski, Skarecky, Bailey and Ayala1994, Reference Kwiatowski, Krawczyk, Jaworski, Skarecky and Ayala1997; Kambysellis et al., Reference Kambysellis, Ho, Craddock, Piano, Parisi and Cohen1995; Russo et al., Reference Russo, Takezaki and Nei1995; Remsen & DeSalle, Reference Remsen and DeSalle1998; Tatarenkov et al., Reference Tatarenkov, Kwiatowski, Skarecky, Barrio and Ayala1999, Reference Tatarenkov, Zurovcova and Ayala2001; Davis et al., Reference Davis, Kurihara, Yoshino and Yamamoto2000 b; Gailey et al., Reference Gailey, Ho, Ohshima, Liu, Eyassu, Washington, Yamamoto and Davis2000; Katoh et al., Reference Katoh, Tamura and Aotsuka2000; Hu & Toda, Reference Hu and Toda2001; Tarrio et al., Reference Tarrio, Rodriguez-Trelles and Ayala2001; Remsen & O'Grady, Reference Remsen and O'Grady2002; Da Lage et al., Reference Da Lage, Kergoat, Maczkowiak, Silvain, Cariou and Lachaise2007; O'Grady & DeSalle, Reference O'Grady and DeSalle2008; van der Linde & Houle, Reference van der Linde and Houle2008). Our results confirm that several genera – Hirtodrosophila, Mycodrosophila, Zaprionus, Scaptomyza and Liodrosophila – are placed between the three major clades of the subgenus Drosophila. The ML and Bayesian analyses differed slightly in the exact placement of the Hirtodrosophila/Mycodrosophila, Zaprionus and immigrans-tripunctata clades, but agreed on all other nodes.
A resolution to the paraphyletic nature of the genus Drosophila will be addressed separately. Three solutions are available: (1) do nothing (O'Grady & Markow, Reference O'Grady and Markow2009); (2) sink the included genera into Drosophila and (3) split the genus along the major clades. Splitting the genus is clearly the most desirable from a purely taxonomic point of view but has the major practical disadvantage that the type specimen for the genus is D. funebris (Fabricius) which is not in the same clade as D. melanogaster. To avoid the widespread confusion that would result from renaming D. melanogaster, an application to preserve the name D. melanogaster has been submitted to the International Commission on Zoological Nomenclature and is currently under consideration (van der Linde et al., Reference van der Linde, Bächli, Toda, Zhang, Katoh, Hu and Spicer2007). If the commission rules in favour of this application, proposals to split the genus are more likely to be entertained by the enormous Drosophila community.
(ix) Impact of analytical approach
Despite the limited character overlap for some species and uneven sampling, our results are remarkably robust across the methods of analysis. Results obtained with different methods show no strong conflict. Many clades are well supported in all of the analyses. In addition, partitioning under ML had little effect on the topology. The partitioned and unpartitioned results differ in only a small number of nodes, and none of these differences received strong support under either partitioning scheme. Partitioned bootstrap values were generally greater than unpartitioned. For example, 45 nodes had greater bootstrap values under partitioning (of four percentage points or more) than without, compared to 14 with the reverse, for those values reported on Fig. 3. Sixty-three (35·6%) nodes differed by no more than three points. The most noticeable differences were between Bayesian and ML support values. As has often been reported, posterior probabilities can be much higher than bootstrap values, sometimes artefactually so (Lewis et al., Reference Lewis, Holder and Holsinger2005 a). The most striking examples in our analyses were in the immigrans-tripunctata radiation, where bootstrap values of less than 40, some as low as 18, were associated with near 100% posterior probabilities. We therefore interpret some of these high pp values with caution.
Considerable debate has surrounded the relative merits of the supermatrix approach as used here and the supertree approach, the latter synthesizing topological information across studies (Bininda-Emonds et al., Reference Bininda-Emonds, Jones, Price, Grenyer, Cardillo, Habib, Purvis and Gittleman2003; Bininda-Emonds, Reference Bininda-Emonds2004; Gatesy et al., Reference Gatesy, Baker and Hayashi2004; de Queiroz & Gatesy, Reference de Queiroz and Gatesy2007). A particular concern about the supermatrix approach is the potential bias created by large numbers of missing data, an issue side-stepped by supertree approaches because the latter do not directly analyse characters. Most studies exploring missing data in large sets have concluded that the supermatrix approach is relatively robust to this problem and that the real question is how many informative data is present, not how many might be missing (Wiens, Reference Wiens2003; de Queiroz & Gatesy, Reference de Queiroz and Gatesy2007; Wiens & Moen, Reference Wiens and Moen2008). Reassuringly, the primary results we report here are well supported and consistent with those revealed by a supertree approach (van der Linde & Houle, Reference van der Linde and Houle2008), a pattern that Baker et al. (Reference Baker, Savolainen, Asmussen-Lange, Chase, Dransfield, Forest, Harley, Uhl and Wilkinson2009) argued was evidence against a misleading bias in either method. We note, however, that the simulation studies have concentrated on the number of missing data, not their distribution within the matrix – in other words, on the behaviour of phylogenetic methods when overlap between some taxa is limited. This focus certainly reduces the effective phylogenetic information below the total number of characters for each taxon. In addition, Lemmon et al. (Reference Lemmon, Brown, Stanger-Hall and Lemmon2009) have shown that ML and Bayesian methods can be positively misleading or provide inflated support values as a result of ambiguous (missing) data. We therefore look forward to future studies that are more complete.
4. Conclusions
Our study includes more drosophilid taxa than any previous molecular phylogenetic study. We obtained better taxon sampling of subfamily Drosophilinae than previous studies by focusing attention on species that are not traditionally assigned to the genus Drosophila, which have been omitted from most previous studies. We obtained this coverage by assembling a matrix of data with a great number of missing data. Despite the potential pitfalls of analyses of such data, results obtained by different methods produced similar results, adequately resolving many aspects of the overall phylogeny, including several long-standing issues. Our study confirms the general observation that the genus Drosophila is paraphyletic and points toward issues that still need attention.
We thank Dr Jean-Luc Da Lage for providing us with the aligned AmyRel data, Clemens Lakner for his help with the parallel version of MrBayes, Dr Jean David for providing us with stocks of several species, Jeff Birdsley for his fly collections and contributions and Dr Anne B. Thistle for her editorial assistance. We also thank the two anonymous reviewers for their constructive comments. This work was supported by National Science Foundation grants DEB-0129219 and the NIH Roadmap for Medical Research, Grant U54 RR021813 to DH and DEB-0454673 and 0841447 to SJS.






