


Materials science has traditionally been driven by scientific intuition followed by experimental study. In recent years, theory and computation have provided a secondary avenue for materials property prediction and design. Several successful examples of materials designed in a computer and then realized in the laboratory Reference Hautier, Jain and Ong1 have now established such methods as a new route for materials discovery and optimization. As computational methods approach maturity, new and complementary techniques based on statistical analysis and machine learning are poised to revolutionize materials science.
While the modern use of the term materials informatics dates back only a decade ago, Reference Rajan and Mendez2 the use of an informatics approach to chemistry and materials science is as old as the periodic table. When Mendeleev grouped together elements by their properties, the electron was yet to be discovered, and the principles of electron configuration and quantum mechanics that underpin chemistry were still many decades away. However, Mendeleev's approach not only resulted in a useful classification but could also make predictions: missing positions in the periodic table indicated potential new elements that were later confirmed experimentally. Mendeleev was also able to spot inaccuracies in atomic weight data of the time. Today, the search for patterns in data remains the goal of materials informatics, although the tools have evolved considerably since Mendeleev's work.
While materials informatics methods are still in their infancy compared to other fields, advancements in materials databases and software in the last decade are rapidly gaining ground. In this paper, we discuss recent developments of materials informatics, concentrating specifically on connecting a material's crystal structure and its composition to its properties (and ignoring, for instance, microstructure and processing). First, we provide a brief history of classic studies based on mining crystallographic databases. Next, we describe the recent introduction of computation-based databases and their potential impact on the field, followed by a discussion of techniques and illustrative examples of modern materials informatics. Finally, we present a novel materials informatics study, based exclusively on openly-available data sets and tools, that predicts the valence and conduction band character of new materials. We note that while this review is mainly focused on periodic solids, molecular systems have also been extensively studied through data mining approaches. Reference Rupp, Proschak and Schneider3–Reference Hansen, Montavon, Biegler, Fazli, Rupp, Scheffler, Von Lilienfeld, Tkatchenko and Müller5
I. EARLY EXAMPLES OF DATA MINING CRYSTALLOGRAPHIC DATABASES
The earliest and still today the most systematic and organized data sets in materials science are based on crystallographic data. Crystal structures of observed compounds are available in databases such as the Inorganic Crystal Database (ICSD), Reference Bergerhoff, Hundt, Sievers and Brown6 the Cambridge Structural Databases, Reference Allen7 and the Pauling file. Reference Villars8 Information on unit cells, atomic positions, and symmetry are available from these resources for hundreds of thousands of inorganic compounds.
Crystal structure data have been extensively used to perform, before the term even existed, data mining studies. For instance, ionic radii for the elements were extracted from large crystallographic data sets in the beginning of the 1970s by Shannon.
Reference Shannon9
This was followed by a more complex description of bonding through the bond valence formalism,
Reference Brown and Altermatt10,Reference O'Keefe and Brese11
which relates the valence of cation i to a sum of the bond strengths s
ij
between cation i and anions j through an analytical expression, i.e., V
i
= ∑
j
s
ij
in which the s
ij
terms are summed over bonds through a simple mathematical expression such as s
ij
= (r
ij
/r
0)−N
or
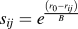 ${s_{ij}} = {e^{{{\left( {{r_0} - {r_{ij}}} \right)} \over B}}}$
. The parameters r
0, N, or B are specific to a cation–anion pair (ij) and must be extracted from a data set through a fitting procedure. Using the ICSD database,
Reference Bergerhoff, Hundt, Sievers and Brown6
Brown et al. extracted these parameters from 750 atom pairs, building a bond valence table that remains widely used today.
Reference Brown and Wu12
${s_{ij}} = {e^{{{\left( {{r_0} - {r_{ij}}} \right)} \over B}}}$
. The parameters r
0, N, or B are specific to a cation–anion pair (ij) and must be extracted from a data set through a fitting procedure. Using the ICSD database,
Reference Bergerhoff, Hundt, Sievers and Brown6
Brown et al. extracted these parameters from 750 atom pairs, building a bond valence table that remains widely used today.
Reference Brown and Wu12
More specific questions on bonding in solids have also been answered through the usage of large structural data sets, e.g., the nature of hydrogen bonds Reference Brown13 and bonding in borates. Reference Yu and Xue14 Furthermore, these data sets have enabled studies of the distribution of inorganic compounds among space groups, Reference Mackay15,Reference Urusov and Nadezhina16 to the search for materials with specific crystallographic and symmetry requirements such as ferroelectrics, Reference Abrahams17 and for screening structure-based properties such as diffusion paths. Reference Avdeev, Sale, Adams and Rao18
Another early application of crystallographic databases was in crystal structure prediction. Early approaches used structure maps, which plot the experimentally observed type of crystal structures against intuitive chemical descriptors (electronegativity, Mendeleev number, or ionic radius). Reference Muller and Roy19–Reference Pettifor22 The groupings that form on these maps can then be used to infer the structure in which a new chemical compound will crystallize. An example of a structure map is plotted in Fig. 1 for the A1 B1 stoichiometry. Recently, Morgan et al. used modern cross-validation techniques to demonstrate that structure maps are indeed predictive and quantified their performances. Reference Morgan, Rodgers and Ceder23 Structure maps have also been combined with modern data mining techniques to build a predictive model using information entropy and classification trees for the prediction of binary halide scintillators. Reference Kong, Luo, Arapan, Villars, Iwata, Ahuja and Rajan24

FIG. 1. An example of a structure map for the A1 B1 composition. Each symbol indicates a specific crystal structure prototype. The axis refers to a “chemical scale” attributing a number to each element based on its position in the periodic table (Mendeleev number). Image from Ref. Reference Pettifor20. ©IOP publishing, all rights reserved. Reprinted with permission.
II. NEW RESOURCES: THE ADVENT OF COMPUTATIONAL MATERIALS DATA REPOSITORIES AND OPEN SOFTWARE
While it is possible to perform data mining using crystal structures alone, most informatics studies additionally require materials property measurements. Although many databases of experimental materials properties are now available, it can be difficult to extract large-scale structure-property relationships from these resources. Computational databases, while also possessing many important limitations, may be able to supplement the capabilities of experimental databases and facilitate an informatics-style approach to materials design.
A. Experimental materials databases
One class of experimental materials databases are the crystallographic structure repositories mentioned previously, which include the ICSD, Reference Bergerhoff, Hundt, Sievers and Brown6 the Pauling file, Reference Villars8 CRYSTMET, Reference White, Rodgers and Le Page25 and Pearson's Crystal Data. Reference Villars and Cenzual26 These resources have been recently summarized and reviewed by Glasser. Reference Glasser27 Materials properties databases are also available. The largest of these is likely the set of databases from Springer, which includes the comprehensive Landolt–Börnstein Database. 28 However, most materials property information remains scattered across multiple resources, including the FactSage suite of databases, Reference Bale, Bélisle, Chartrand, Decterov, Eriksson, Hack, Jung, Kang, Melançon, Pelton, Robelin and Petersen29 the National Institute of Standards and Technology databases, Reference Linstrom and Mallard30 MatWeb, 31 MatNavi, 32 various publications such as the Kubaschewski tables, Reference Kubaschewski, Alcock and Spencer33 and the Handbook of Ternary Alloy Phase Diagrams. Reference Okamoto, Villars, Prince and Okamoto34 We note that Citrine Informatics (http://www.citrine.io) is one commercial entity that is attempting to centralize information collected from diverse sources (both experimental and computational).
These various data sources have historically been expertly curated and validated, and serve as important, trusted resources for the materials research community. While it is certainly possible to perform data mining on these databases, limitations include completeness and programmatic access. In terms of completeness, many materials properties (e.g., formation energies, band gaps, and elastic tensors) have only been measured for only a small fraction of the number of known crystal structures. There is particularly a lack of data available for negative results, including failed synthesis attempts and unexceptional materials properties measurements. Even when compound properties are available, they are often associated only with a composition and lack a rigorous description of the material being measured (crystal structure, microstructure, doping level, etc.). The lack of information about the input material can make it very challenging to develop models. Finally, in terms of data access, most of these databases can only be accessed through mechanisms designed for “single lookups” rather than systematic data mining over large portions of the database. Thus, there is room for other types of databases that can help address the gaps in the experimental record.
B. Computational materials databases
In recent years, the ability to generate materials data using systematic high-throughput computations (typically based on density functional theory, or DFT, approaches for solving the Schrödinger equation Reference Hohenberg and Kohn35,Reference Kohn and Sham36 ) has created new, efficient opportunities to produce high-quality databases for data mining. These computationally-driven databases, which usually leverage crystal structure information from experimental databases, provide powerful means to extract patterns and correlations from hitherto unavailable data sets. As an example, the full elastic tensor has only been measured for approximately 150 distinct compounds, but a recent high-throughput computational study has tabulated this quantity for over 1000 materials. Reference Jong, Chen, Angsten, Jain, Notestine, Gamst, Sluiter, Ande, Zwaag, Plata, Toher, Curtarolo, Ceder, Persson and Asta37
Examples of such computationally-derived databases include the Materials Project, Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 AFLOWlib, Reference Curtarolo, Setyawan, Wang, Xue, Yang, Taylor, Nelson, Hart, Sanvito, Buongiorno-Nardelli, Mingo and Levy39 the Open Quantum Materials Database, Reference Saal, Kirklin, Aykol, Meredig and Wolverton40 the Harvard Clean Energy Project, Reference Hachmann, Olivares-Amaya, Atahan-Evrenk, Amador-Bedolla, Sanchez-Carrera, Gold-Parker, Vogt, Brockway and Aspuru-Guzik41 the Electronic Structure Project, Reference Ortiz, Eriksson and Klintenberg42 NoMaD, Reference Blokhin, Pardini, Mohamed, Hannewald, Ghiringhelli, Pavone, Carbogno, Freytag, Draxl and Scheffler43 NRELMatDB, Reference Stevanović, Lany, Zhang and Zunger44 and the Computational Materials Repository. Reference Landis, Hummelshøj, Nestorov, Greeley, Dulak, Bligaard, Norskov and Jacobsen45 Some of these databases can be quite extensive; for example, the Materials Project Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 today contains property data for over 60,000 compounds and includes many different properties, and AFLOWlib Reference Curtarolo, Setyawan, Wang, Xue, Yang, Taylor, Nelson, Hart, Sanvito, Buongiorno-Nardelli, Mingo and Levy39 includes over 600,000 entries. However, more focused efforts are also proliferating, including CatApp Reference Hummelshøj, Abild-Pedersen, Studt, Bligaard and Nørskov46 for catalysis, PhononDB Reference Togo and Tanaka47,Reference Togo48 for phonons, TEDesignLab for thermoelectrics, Reference Gorai, Gao, Ortiz, Miller, Barnett, Mason, Lv, Stevanović and Toberer49 and ESTEST Reference Yuan and Gygi50 for verification and validation of physics software. In some cases, the separation between these efforts is clear. For example, the Harvard Clean Energy Project Reference Hachmann, Olivares-Amaya, Atahan-Evrenk, Amador-Bedolla, Sanchez-Carrera, Gold-Parker, Vogt, Brockway and Aspuru-Guzik41 is geared toward small molecules, whereas AFLOWlib Reference Curtarolo, Setyawan, Wang, Xue, Yang, Taylor, Nelson, Hart, Sanvito, Buongiorno-Nardelli, Mingo and Levy39 targets inorganic compounds. In other cases, such as for the Materials Project, Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 AFLOWlib, Reference Curtarolo, Setyawan, Wang, Xue, Yang, Taylor, Nelson, Hart, Sanvito, Buongiorno-Nardelli, Mingo and Levy39 and the Open Quantum Materials Database, Reference Saal, Kirklin, Aykol, Meredig and Wolverton40 there is considerable overlap in the intended scope. Even in this latter case, users can still benefit from multiple databases, e.g., to verify results or to “fill in the gaps” of their preferred database. Unfortunately, at present there exists no search-engine or similar tool to facilitate search across databases (e.g., something in the spirit of ChemSpider Reference Pence and Williams51 ). This might partly be due to the current difficulty of accessing data programmatically in many of these resources, as discussed in Sec. II. C. A summary and comparison of these different efforts can be found in a recent review by Lin. Reference Lin52
A major contributing factor to the rise of simulation-based data is the availability of software libraries that have brought large-scale data generation and data mining within the reach of a greater number of research groups. Examples include pymatgen Reference Ong, Richards, Jain, Hautier, Kocher, Cholia, Gunter, Chevrier, Persson and Ceder53 (materials analysis, plotting, and I/O to DFT software), ASE Reference Bahn and Jacobsen54 (structure manipulation and DFT calculator interface), AFLOW Reference Curtarolo, Setyawan, Hart, Jahnatek, Chepulskii, Taylor, Wang, Xue, Yang, Levy, Mehl, Stokes, Demchenko and Morgan55 (high-throughput DFT framework), AiiDA Reference Pizzi, Cepellotti, Sabatini, Marzari and Kozinsky56 (workflow management for high-throughput DFT), and FireWorks Reference Jain, Ong, Chen, Medasani, Qu, Kocher, Brafman, Petretto, Rignanese, Hautier, Gunter and Persson57 (general workflow software for high-throughput computing). These codebases, as well as the continually improving accuracy of theoretical techniques, robust and more powerful DFT software, and the exponential growth of computing power will likely make simulation-based data sets even more valuable and prevalent in the future.
C. Programmatic data access
An efficient method to download large data sets from data resources (whether experimental or computational) is necessary for performing materials informatics. There are many methods by which data can be exposed, including direct download of either raw or processed data sets. A more modern technique to expose a data resource is to use representational state transfer (REST) principles to create an application programming interface (API) to the database. Reference Fielding58 This method was pioneered in the computer science community and was introduced to the materials world through the materials API (MAPI) Reference Ong, Cholia, Jain, Brafman, Gunter, Ceder and Persson59 of the Materials Project. To date, the MAPI has served more than 15 million pieces of materials data for over 300 distinct users, enabling new types of applications and analyses.
Under RESTful design, each object is represented as a unique resource identifier (URI) that can be queried in a uniform manner using the hypertext transfer protocol (HTTP). Each document or object (such as a computational task, crystal structure, or material property) is represented by a unique URI (see Fig. 2 for an example) and an HTTP verb that can act on that object. In most cases, this action returns structured data that represents the object, e.g., in the javascript object notation format (JSON).

FIG. 2. An example of the URL structure of the MAPI. Figure reprinted from Ref. Reference Ong, Cholia, Jain, Brafman, Gunter, Ceder and Persson59 with permission from Elsevier.
Some of the advantages of RESTful interfaces include:
-
(i) Abstraction: RESTful interfaces use universal protocols that can be accessed by many programming languages. They hide the details of the underlying data storage implementation (i.e., whether the data is stored in an SQL or NoSQL database), by exposing a clean and consistent set of actions and queries that can be performed against the data.
-
(ii) Flexibility: Because they abstract away implementation details, RESTful interfaces are flexible to changes in the underlying infrastructure. They also allow for federation amongst several databases with different internal architectures under a consistent API, such that a user can in principle write the same code for different resources. Such flexibility might become especially important in building universal access modes to disparate data sources.
-
(iii) Power: High-level interfaces can be built upon RESTful APIs such that one can access and manipulate offsite data resources in an object-oriented way. For example, a high-level interface to the MAPI Reference Ong, Cholia, Jain, Brafman, Gunter, Ceder and Persson59 is provided in the pymatgen Reference Ong, Richards, Jain, Hautier, Kocher, Cholia, Gunter, Chevrier, Persson and Ceder53 code base and allows users to obtain properties, such as crystal structure or electronic band structure, using built-in functions rather than by explicitly making HTTP requests.
-
(iv) Up-to-date: Data sets can become stale and outdated. A RESTful interface allows for the most recent version of the data and queries to be exposed at all times without actions needed by the user. Users can always choose to retain data, and the URI scheme can also be used to preserve multiple versions of the data. However, RESTful APIs make it simple to obtain the most current and relevant data for a given analysis without needing to re-download the entire database.
Although REST interfaces can be tricky for beginning users, a well-designed REST interface promotes discoverability of the data and frees the end user from learning the implementation details of a particular database, instead allowing data analysis through a clean and consistent API.
III. MODERN DATA MINING TECHNIQUES AND EXAMPLES
With the generation of ever-expanding materials data sets underway, the major remaining challenges are to develop descriptors (sometimes called “features” or “predictors”) of materials and relate them to measured properties (sometimes called “outputs” or “responses”) through an appropriate data mining algorithm. In the last few decades, many new approaches have been developed to extract knowledge from large data sets using elaborate mathematical algorithms, leading to the new field of machine learning or data mining. Reference Hastie, Tibshirani and Friedman60 In many cases, such algorithms can be applied in an “off-the-shelf” way for materials problems; in other cases, materials scientists have themselves developed new approaches for data analysis that are tuned to their domain.
A. Descriptors for materials structure and properties
Because data mining operates on numerical data structures, materials scientists must first encode materials in a format that is amenable for finding relationships in the data. While several data formats have been developed for describing crystallographic materials (e.g., the CIF file format), these formats are not suitable as data mining descriptors for reasons that follow. The problem of developing robust descriptors for crystalline solids remains a challenging task; here, we identify and recapitulate Reference Ghiringhelli, Vybiral, Levchenko, Draxl and Scheffler61 four properties that characterize good descriptors:
-
(i) Descriptors should be meaningful, such that relationships between descriptors and responses are not overly complex. For example, whereas the lattice vectors and atom positions of a crystal structure in principle determines its properties, such an encoding involves a very complex relationship between inputs and outputs (i.e., the Schrödinger equation). In particular, the complex and important three-dimensional boundary conditions implicit in this representation are not captured by today's data mining techniques. Better descriptors have simpler relationships to the outputs, ideally within the complexity space of what a data mining algorithm can reasonably uncover (in accordance with similar principles outlined by Ghiringhelli et al. Reference Ghiringhelli, Vybiral, Levchenko, Draxl and Scheffler61 )
-
(ii) Better descriptors are universal, such that they can be applied to any existing or hypothetical material. While this is not strictly necessary when performing analysis within a well-constrained chemical space, it is useful for building universal models that bridge chemistries and structures.
-
(iii) Better descriptors are reversible, such that a list of descriptors can in principle be reversed back into a description of the material. This is not strictly necessary for a successful model, but it would enable more efficient “inverse design” in descriptor space rather than in the space of materials. A less stringent version of this condition was put forth by Ghiringhelli et al., Reference Ghiringhelli, Vybiral, Levchenko, Draxl and Scheffler61 who stated that descriptors should uniquely characterize a material.
-
(iv) Descriptors should be readily available, i.e., they should be easier to obtain than the target property being predicted. Reference Ghiringhelli, Vybiral, Levchenko, Draxl and Scheffler61
It is unlikely that any one set of descriptors will meet such criteria across the space of all potential compositions, crystal structures, and targeted output properties. Rather, descriptors will likely need to be tailored to an application, as was demonstrated by Yang et al. for topological insulators. Reference Yang, Setyawan, Wang, Buongiorno Nardelli and Curtarolo62
A common first-level set of descriptors is to encode compounds as a vector that depends only on the identity of the elements contained within the compound, without explicit consideration of crystal structure or stoichiometry. For example, an A–B binary compound might include properties such as the electronegativity of the pure A element, the electronegativity of the pure B element, the atomic radii of these elements, and the experimentally known melting points of these elements. One weakness of this strategy is that the properties of pure elements do not always correlate well to their properties exhibited in compounds; as an extreme example, the elemental properties of oxygen gas are very different from the anionic properties of oxygen in oxide compounds. An extension of this basic technique, which we refer to as the “atoms-in-compounds” approach, still only considers the identity of the atoms in a compound when formulating descriptors, but uses descriptions of the elements from known compound data. For example, for the same A–B compound we might include descriptors such as the ionic radius of A (determined from its bonds in the context of selected compounds) or the oxide heat of formation of B. Depending on the type of compound being modeled, such descriptors might be more predictive than properties of the pure elements.
Important research efforts are being directed toward the development of novel descriptors today for encoding more complex materials data such as crystal structure. For instance, beyond the description of materials by space groups, a description by crystal structure prototypes can be essential and several algorithms have been developed Reference Burzlaff and Zimmermann63,Reference Allmann and Hinek64 and applied toward probabilistic crystal structure prediction models. Reference Hautier, Fischer, Jain, Mueller and Ceder65,Reference Hautier, Fischer, Ehrlacher, Jain and Ceder66 Further methods include the development of the “symmetry functions” that capture two and three-body terms Reference Behler and Parrinello67 and local environment descriptors Reference Yang, Dacek and Ceder68 to characterize bonding details in crystals. “Fingerprints” representing the somewhat complex object of crystal structures have also been proposed to characterize the landscape of possible structures and as descriptors in data mining approaches. Reference Oganov and Valle69,Reference Isayev, Fourches, Muratov, Oses, Rasch, Tropsha and Curtarolo70 Finally, other recent approaches are inspired by molecular data mining; the idea of a “coulomb matrix” as a representation of organic molecules has been extended to solids by Faber et al., Reference Faber, Lindmaa, von Lilienfeld and Armiento71 and the SimRS model for structure primitives has been applied to solids by Isayev et al. Reference Isayev, Fourches, Muratov, Oses, Rasch, Tropsha and Curtarolo70
In addition to descriptors derived from experimental data and structural analysis, descriptors from DFT computations are becoming increasingly popular. For instance, a data mining approach using linear regression recently revealed that information from the charge density of the material could be predictive of its elastic tensor. Reference Kong, Broderick, Jones, Loyola, Eberhart and Rajan72 Mixing of computed and traditional descriptors is also possible; for example, Seko et al. Reference Seko, Maekawa, Tsuda and Tanaka73 presented results on the development of a linear regression model predicting melting temperature from both classical descriptors and computed quantities (such as cohesive energies). This study highlights the gain in predictive power provided by the addition of these computed quantities (see Fig. 3), provided that performing the computations remains more convenient than determining the final property. We note that even properties for which the underlying physics is unclear, such as high-T c superconductors, can sometimes be tackled by combining computed data (density of states (DOS) and band structures) with experimental observations. Reference Isayev, Fourches, Muratov, Oses, Rasch, Tropsha and Curtarolo70

FIG. 3. Predicted versus experimental melting temperature over a data set of 248 compounds for two models: one including DFT descriptors and one without. Image from Ref. Reference Seko, Maekawa, Tsuda and Tanaka73. Reprinted with permission, ©American Physical Society.
Often, such fundamental descriptors perform better if they are combined through functions; for example, a descriptor describing the ratio between the ionic radius of elements A and B in a compound may produce more accurate linear models than two separate descriptors describing the individual radii. We note that functions that are symmetric to the interchange of elements can be used to avoid the problem of site differentiation across crystal structures. One problem in formulating such functions, and with descriptor selection in general, is the large number of possibilities and the danger of using correlated predictors, and in over-fitting. An active area of research in statistical learning methods is in such feature selection problems, which identify the best descriptors within a large pool. For example, recent work by Ghiringelli et al. Reference Ghiringhelli, Vybiral, Levchenko, Draxl and Scheffler61 have demonstrated that physically relevant models can be constructed by mathematically selecting between approximately 10,000 possible functional forms of descriptor combinations. Such techniques to automatically derive physical relations are reminiscent of earlier work in physics data mining, in which the equations of motion of a double pendulum could be automatically derived given explicit input variables and an output data set. Reference Schmidt and Lipson74
B. Exploratory data analysis and statistics
Once the descriptors and target outputs are determined, an appropriate data mining scheme must be selected to relate these quantities. The first step is to plot visual correlations and apply standard statistical tools (such as the analysis of variance, or ANOVA, approach) to better understand the data set and to make basic predictions. This phase is often referred to as exploratory data analysis. For example, one large ab initio data set Reference Jain, Hautier, Moore, Ong, Fischer, Mueller, Persson, Ceder and Ping Ong75 targeted the discovery of new Li-ion cathode materials. This data set encompassed tens of thousands of materials, which were extensively analyzed for trends, limits, and opportunities across cathode chemistries. For example, statistics for this data set were compiled for phosphate materials, Reference Hautier, Jain, Ong, Kang, Moore, Doe and Ceder76 which have been of great interest for the battery community due to the success of LiFePO4 as a Li-ion cathode. Using more than 4000 lithium-containing phosphate compounds, the expected voltage for all potential redox couples in any phosphate cathode material could be derived and rationalized. Furthermore, several hypotheses, proposed in the literature and based on limited data, on the relevant factors determining voltage (such as ratio of P/O, number of linkages between phosphate groups and redox metals, and P–O bond length) were confirmed or challenged.
Visual exploration can also uncover relationships between properties. The same project mentioned above uncovered a correlation between the computed average voltage and thermodynamic chemical potential of O2 (translated into a temperature for oxygen gas release) for cathode compounds (Fig. 4). Reference Jain, Hautier, Ong, Dacek and Ceder77,Reference Ong, Jain, Hautier, Kang and Ceder78 This analysis determined that higher voltage compounds are in general at greater risk for thermal instability. Although a similar idea was proposed much earlier, Reference Godshall, Raistrick and Huggins79 the advent of high-throughput computed data tested the idea on a large scale and further uncovered chemistry-based differences in behavior.

FIG. 4. Temperature of O2 release versus voltage for a large set of cathode materials. Higher temperatures are associated with greater “safety” of the cathode material. A clear correlation between higher voltage and lower temperatures for releasing oxygen gas is observed. The figure on the right depicts a linear least-squares regression fit to the data for different chemistries (oxides, sulfates, borates, etc.). While all cathode materials have a similar tendency to be less safe for higher voltage, there is a clear difference between different (poly)anions. Image from Ref. Reference Jain, Hautier, Ong, Dacek and Ceder77. Reproduced by permission of the PCCP Owner Societies.
C. Clustering
Going beyond visual examination and basic correlation analysis, one can use clustering techniques to uncover hidden relationships in the data. Clustering divides data into groups based on a similarity metric in a way that uncovers patterns and categories, but does not directly predict new values. Several algorithms exist today for automatically clustering data (e.g., k-means clustering, Ward hierarchical clustering, Density-based spatial clustering of applications with noise (DBSCAN), and others Reference Xu and Wunsch II80,Reference Gan, Ma and Wu81 ). To perform a clustering analysis, one must be able to define a distance metric between data points (e.g., Manhattan distance or Euclidean distance between feature vectors) and sometimes additional parameters depending on the algorithm (e.g., a standard k-means clustering requires setting the number of clusters in advance); several different trials of an iterative algorithm may be needed to find an optimal grouping.
The results of a clustering analysis can be used downstream in data analysis; for example, each cluster might represent a “category” of material, and a separate predictive model can be fit for each cluster. For example, this is the approach taken in the cluster-rank-model method developed by Meredig and Wolverton, Reference Meredig and Wolverton82 which was used to classify and then predict the stability of various dopants into zirconia.
One technique for visualizing a hierarchical clustering process is the dendrogram, which displays the results of clustering as a function of cutoff distance. An example of a dendrogram for two dimensions of clustering is depicted in Fig. 5. This diagram was created by Castelli and Jacobsen Reference Castelli and Jacobsen83 to understand which elements can be placed in the A and B sites of a perovskite structure to produce a stable compound that also has a nonzero band gap, with the end goal being to understand principles for designing photocatalyst materials. The researchers observed several “pockets” representing combinations of elements that met their criteria (Fig. 5) and that could be rationalized through the periodic table. The way to read the dendrogram tree in Fig. 5 is as follows:
-
(i) The dendrogram can be cut at any distance; the number of lines intersecting that cut is the number of clusters formed at that distance cutoff.
-
(ii) As one progressed down the tree (reduces the distance cutoff), more clusters emerge representing groups of greater homogeneity. For example, in terms of the A ion (right side of Fig. 5), the first “cut” separates La from the other elements, and the second cut separates Y; further cuts divide the remaining elements into groups. In practice, such hierarchical clustering algorithms can either be agglomerative (start with each data point as a separate cluster and begin merging them) or divisive (start with all data points in a single cluster, and start dividing those clusters).
-
(iii) By coloring the leaf nodes, the distinct clusters at a selected cutoff can be emphasized.

FIG. 5. Elements forming stable oxide perovskites in the A and B sites; the gap is represented by the color, and elements are ordered by size of gap and grouped by similarity in gap. The dendrogram trees for A and B sites are plotted at the right and top of the image, respectively. Image from Ref. Reference Castelli and Jacobsen83. ©IOP publishing, all rights reserved. Reprinted with permission.
Clustering is often combined with methods for dimension reduction such as principal components analysis, which determines orthogonal directions in high-dimensional space that explain the most variance in the data. Often, only a few principal components are needed to explain most of the data variance, allowing clustering to be performed in a small number of dimensions (e.g., 2 or 3) and subsequently visualized. One example of such a clustering analysis is the classification of oxide DOS spectra performed by Broderick et al. Reference Broderick, Aourag and Rajan84 In this study, DFT-based DOS data for 13 compounds were normalized and aligned such that each DOS was parametrized as a 1000 element vector relating energies to number of states. A principal components analysis was then applied to determine which parts of the DOS explained the greatest difference between materials; i.e., the first principle component, which is itself a 1000-element vector resembling a DOS, was highly related to the average difference in the DOS spectrum between monoclinic and cubic/tetragonal structures. Each DOS in the data set was then projected in the space of the first two principle components, and a clustering analysis was performed to find similarities in this space. By examining the clusters, it was determined that variation in the (normalized, aligned) DOS was most affected in the order structure >stoichiometry >chemistry, although it should be noted that there exist hidden relationships between these variables in the data.
A larger-scale version of DOS classification was performed by Isayev et al., Reference Isayev, Fourches, Muratov, Oses, Rasch, Tropsha and Curtarolo70 who grouped together structures in the AFLOWlib Reference Curtarolo, Setyawan, Wang, Xue, Yang, Taylor, Nelson, Hart, Sanvito, Buongiorno-Nardelli, Mingo and Levy39 database through the use of a “D-fingerprint” to encode DOS information (similar to the encoding used by Broderick et al. Reference Broderick, Aourag and Rajan84 described previously). Rather than assessing similarity in the space of principal components, the distance between these D-fingerprint vectors was computed directly and used to construct force-directed graphs that placed materials with similar DOS together. While the resulting data set is complex, some of the major groups that emerged were those with a similar number of distinct elements in the compound as well as a similar number of atoms in the unit cell. The authors of the study used such graphs only for making qualitative statements, and explored other methods for performing quantitative analysis.
D. Linear models
When quantitative predictions are required, one option is to construct linear models, in which the response value is calculated as some linear combination of descriptor values. Linear models encompass a broad range of techniques and can often be modified to tackle different types of problems. For example, when predicting probabilities Pr(X) with possible values strictly in the interval (0,1), a limitation of standard linear models is that values outside the interval can be predicted. However, by transforming probabilities to a “log-odds” formalism (log[Pr(X)/(1−Pr(X)]) in logistic regression, the interval of prediction can be reformulated into (−∞, +∞). If factor variables (i.e., categorical variables such as {“metal”, “semiconductor”, “insulator”}) are involved, linear models can be used by encoding the potential factor levels as a set of binary coefficients. As a final example, “robust linear models” are resistant to outliers and violations of least-squares linear model assumptions (such as the presence of heteroscedasticity). Reference Andersen85
Linear methods can be powerful methods for estimation and prediction, particularly if descriptors are well-chosen. For example, Chelikowsky and Anderson Reference Chelikowsky and Anderson86 determined that the melting points of 500 intermetallic alloys was highly correlated with a simple average of their (known) unary end-member melting points; however, other descriptors (such as differences in Pauling electronegativity) yielded only weak correlations.
One strategy to find good descriptors for linear models is to begin by including many possibilities and subsequently filter out those that are nonpredictive or redundant. For example, the method of partial least-squares fitting can reduce the effect of collinear descriptors (e.g., components of spectra in which neighboring data points are likely to be highly related). Another technique is regularization of the linear coefficients (i.e., reducing the l 1 and/or l 2 norm) through approaches such as least absolute shrinkage and selection operator (LASSO) Reference Tibshirani87 (penalizes l 1 norm), ridge regression (penalizes l 2 norm), or Elastic net Reference Zou and Hastie88 (penalizes a combination of l 1 and l 2 norm). These methodologies help reduce the number and strength of correlated or unhelpful predictors and have been successfully applied to several materials predictions problems such as chalcopyrite band gap prediction, Reference Dey, Bible, Datta, Broderick, Jasinski, Sunkara, Menon and Rajan89 band gap engineering, Reference Srinivasan and Rajan90 scintillator discovery, Reference Kong and Rajan91 mechanical properties of alloys Reference Toda-Caraballo, Galindo-Nava and Rivera-Díaz-Del-Castillo92 and phosphor data mining. Reference Park, Singh, Kim and Sohn93
An example of the application of such techniques is the use of principal component linear regression analysis by Curtarolo et al. to predict energies of compounds. Reference Curtarolo, Morgan, Persson, Rodgers and Ceder94 In this study, the energy of a compound was demonstrated to be predictable by linear regression from the energies of other compounds in the same chemical system. Another example pertains to new piezoelectrics discovery: Balachandran et al. reduced a set of 30 candidate descriptors down to 6 using principal components analysis. Reference Balachandran, Broderick and Rajan95 This reduced set of descriptors was subsequently incorporated into a linear model that predicted the Curie temperature at the morphotropic phase boundary, including two new possible piezoelectric materials (see Fig. 6).

FIG. 6. Predicted versus experimental Curie temperature for a series of piezoelectric materials. Blue points are the training set, green triangle the test set and red cross predicted new piezoelectrics. Image from Ref. Reference Balachandran, Broderick and Rajan95. ©The Royal Society, reprinted with permission.
E. Kernel ridge regression (KRR)
Linear techniques might not adequately capture complex relationships in the data. One method to capture nonlinear relationships is to retain linear models, but to transform the descriptors in nonlinear ways (e.g., by taking the square or logarithm of descriptors) prior to fitting the linear model. This idea forms the essence of KRR, which leverages two principles beyond standard linear regression. First, KRR uses the ridge regression principle of regularizing the fitted coefficients by adding a penalty term that scales with their l 2 norm. The regularization term, used to control over-fitting, is particularly important for kernel-based methods like KRR because of the higher number of dimensions being used for the fit. The second principle of KRR is to use a kernel to lift the input descriptors into a higher dimensional space. The mathematics of kernels can be found elsewhere, Reference Cristianini and Shawe-Taylor96 but the general principle is that problems that can be posed as inner products can be mapped to higher dimensions through kernels that transform the inputs. This allows KRR to efficiently minimize the ridge regression error function even when the number of dimensions in the transformed feature vectors is very high and perhaps even in excess of the number of observations. There are many types of kernels that can be used for this higher-dimensional mapping, including polynomial, gaussian, exponential, and others. The choice of kernel function and the magnitude of the regularization parameter will affect the fitting and must often be tuned, e.g., through cross-validation.
The KRR model is nonparametric, i.e., the prediction for a new feature vector is not expressed through a typical linear equation expressed in terms of transformed features but rather by projecting that targeted feature vector onto the solution space through dot products with the training data. KRR has been used to model the properties of small molecules (atomization energies Reference Rupp, Tkatchenko, Müller, Lilienfeld and Anatole4 and multiple properties of 1D chains Reference Pilania, Wang, Jiang, Rajasekaran and Ramprasad97 ) and have recently been applied to solids. For example, Shütt et al. used KRR to predict the DOS at the Fermi energy using a sample of 7000 compounds, finding a high correlation (error of roughly 6% of the range of values) that depends on the types of electronic orbitals involved. Reference Schütt, Glawe, Brockherde, Sanna, Müller and Gross98 A second example is from Faber et al., who used KRR to predict formation energies using a sample of almost 4000 compounds, achieving errors as low as 0.37 eV/atom. Reference Faber, Lindmaa, von Lilienfeld and Armiento71 In both instances, a critical part of the analysis was deciding how to represent crystalline materials through descriptors; Shütt et al. leveraged a form similar to radial distribution functions, Reference Schütt, Glawe, Brockherde, Sanna, Müller and Gross98 whereas Faber et al. modified a Coulomb matrix form that was developed for molecules. Reference Faber, Lindmaa, von Lilienfeld and Armiento71
F. Nonlinear techniques based on trees
There exist several methods designed to directly capture nonlinear functions of descriptors that produce an output. One such method is that of neural networks, in which descriptor values serve as “input nodes” that form a weighted graph through “hidden nodes” and end in a set of “output” nodes, with the weightings describing (in a nonlinear way) how the inputs relate to the outputs and are trained by the data. Neural nets have been applied in materials science Reference Jalem, Nakayama and Kasuga99–Reference Zhang, Yang and Evans102 and are experiencing a general resurgence in the form of “deep learning”; however, they suffer from a lack of interpretability, and large data sets are usually needed to adequately train them.
An alternative nonlinear technique is that of tree-based models. Tree-based methods iteratively split variables into successively smaller groups, typically in a way that maximizes the homogeneity within each branch. In this way, trees are similar to clustering, but they are supervised (split on the value of a known “response” variable but using decisions only on the “predictors”) whereas clustering is unsupervised (finds internal relationships between all variables, without distinction between “predictor” and “response”). A tree model makes a series of decisions based on descriptor values that successively move down the branches, until one reaches a “leaf” (ending) node within which the values of the responses are sufficiently homogeneous to make a prediction.
One field in which tree-based methods have been applied is thermoelectrics design. Carrete et al. have constructed decision trees Reference Carrete, Mingo, Wang and Curtarolo103 that separate half-heusler structures into high and low probability of possessing high figure-of-merit based on decisions regarding electronegativity difference, atomic number difference, and periodic table column (see Fig. 7). This study also used the tree-based method of random forests (described next) to predict thermal conductivity.

FIG. 7. Results from a decision tree algorithm on a data set of 75 half heusler compounds; labels above the arrows represent decisions, and nodes are divided into number of compounds and fraction of compounds remaining that possess high ZT (the thermoelectric figure-of-merit). The decision tree highlights the most important factors leading to high ZT materials for two different operating temperatures (300 and 1000 K). Image from Ref. Reference Carrete, Mingo, Wang and Curtarolo103. ©Wiley-VCH Verlag GmbH & Co.,reprinted with permission.
There exist many techniques for improving the predictive performance of trees, such as pruning (removing endpoint branches) to avoid over-fitting and smoothing to reduce variance in prediction (so that small changes in input data do not result in abrupt changes to the output). In recent years, ensemble-based techniques based on averaging the predictions of several models have gained much popularity. For example, the technique of random forests Reference Liaw and Wiener104 fits many individual tree models (as the name suggests) and then averages or takes a vote on the results. To prevent each tree in the random forest from producing the same result, each model is (i) fit using only a subset of the training data and (ii) employs only a subset of the possible descriptors. We note that when only (i) is used, the technique is termed bagging and falls under the general technique of bootstrapping. The random forest method results in a mix of slightly different “opinions” that are used to form a coherent consensus that reduce bias of a single model and produces smoother results than a single tree. We note that ensembling techniques are more general than tree-based methods and can indeed mix many different types of models. For example, Meredig et al. Reference Meredig, Agrawal, Kirklin, Saal, Doak, Thompson, Zhang, Choudhary and Wolverton105 combined a heuristic approach (based on relating ternary formation energies to a weighted sum of binary formation energies) with a regression approach based on ensembles of decision trees to build a model linking composition to formation energy.
Ensembles are effective because a majority vote of many models can produce better results than any individual model, provided that the errors in the individual models are not highly correlated. For example, by combining 5 different models that are each only 75% accurate in predicting a binary outcome (with random errors) into a majority vote ensemble, the overall probability of a correct classification increases to approximately 90%. Unfortunately, such models can be difficult to interpret and might be overfit if a proper machine learning design (training, validation, test sets) Reference Bell, Koren and Volinsky106 is not followed. Nevertheless, the high level of performance that can be achieved will likely make ensembles popular in future materials science studies.
G. Recommendation engines
Another class of data mining techniques is based on finding associations in data and can be used to create a ranked list of suggestions or probable outcomes. Using these techniques, one can derive useful associations in materials properties in the same way that a retailer can predict that a shopper who purchases cereal is likely to also purchase milk.
Indeed, some of the first applications of advanced machine learning algorithms in materials science were based on this idea. Fischer et al. developed a Bayesian statistics-based algorithm trained on a large crystallographic experimental database (the Pauling file Reference Villars8 ) to build correlations between existing binary crystal structures and proposed ones. Reference Fischer, Tibbetts, Morgan and Ceder107 The model used the associations to build predictive models, such as: if A1B1 composition of two elements is known to form the rock salt structure, then what is the probability that a new proposed A2B1 compound of those elements crystallizes in the Mg2Si prototype structure? Fischer et al. analyzed such associations to derive probabilistic statements on what structures are likely to form at unexplored compositions based on what structures are observed at measured compositions. This approach was later extended to ternary systems and in particular ternary oxides to perform an automatic search for new compounds. Reference Hautier, Fischer, Jain, Mueller and Ceder65 Among the reported successes is the experimental confirmation of the formation of SnTiO3 in an ilmenite structure rather than perovskite (as was previously proposed in the literature Reference Fix, Sahonta, Garcia, MacManus-Driscoll and Blamire108 ).
Subsequently, another compound prediction data mining-based algorithm, specifically designed for data-sparse regions such as quaternary compounds, was proposed by Hautier et al. Reference Hautier, Fischer, Ehrlacher, Jain and Ceder66 This method is based on the simple assumption that if a compound forms in a given crystal structure (e.g., a perovskite), replacing one of its ions by an ion that is chemically similar (e.g., in the same column of the periodic table or in the same size) is likely to lead to a new stable compound in the same structure. The idea was implemented within a rigorous mathematical framework and trained on compounds present in the ICSD database. Reference Bergerhoff, Hundt, Sievers and Brown6 Fig. 8 represents a map of pair correlations between ions obtained from the probabilistic model. The map indicates which ions are likely (red) and unlikely (blue) to substitute for one another. Some compounds predicted by this work (mainly rooted in an interest in the Li-ion battery field) and confirmed experimentally include: Li9V3(P2O7)3PO4)2, Reference Hautier, Jain, Ong, Kang, Moore, Doe and Ceder76,Reference Jain, Hautier, Moore, Kang, Lee, Chen, Twu and Ceder109,Reference Kuang, Xu, Zhao, Chen and Chen110 A3M(CO3) (PO4) (A = alkali, M = TM), Reference Chen, Hautier and Ceder111,Reference Hautier, Jain, Chen, Moore, Ong and Ceder112 LiCoPO4, Reference Jähne, Neef, Koo, Meyer and Klingeler113 Li3CuPO4, Reference Snyder, Raguž, Hoffbauer, Glaum, Ehrenberg and Herklotz114 and LiCr4(PO4)3. Reference Mosymow, Glaum and Kremer115 An online version of this tool is available as the Materials Project “Structure Predictor” app. More recent work by Yang et al. has further built on this concept and proposed a composition similarity mapping. Reference Yang and Ceder116

FIG. 8. Map of the pair correlation for two ions to substitute within the ICSD database. Positive values indicate a tendency to substitute, whereas negative values indicate a tendency to not substitute. The symmetry of the pair correlation (g ab = g ba) is reflected in the symmetry of the matrix. Image from Ref. Reference Hautier, Fischer, Ehrlacher, Jain and Ceder66. ©The American Chemical Society, reprinted with permission.
Another example of associative learning is in the prediction of photocatalyst materials. Castelli et al. computed several key photocatalytic properties using DFT for over 18,000 ABO3 oxides, then used association techniques to derive the probability of an arbitrary oxynitride (i.e., ABO2N) to meet the same photocatalytic criteria. Reference Castelli and Jacobsen83 By learning the A and B element identities, most associated with good photocatalytic behavior in the oxides data and transferring the same assumptions for oxynitrides, they were able to find 88% of the target oxynitride systems by searching only 1% of the data set.
Such analyses are culminating in more general “recommender” systems that can suggest new compounds based on observed data. For example, Citrine Informatics has built a recommendation system for suggesting new and unconventional thermoelectric materials. Reference Gaultois, Oliynyk, Mar, Sparks, Mulholland and Meredig117 Another example is that of Seko et al., Reference Seko, Maekawa, Tsuda and Tanaka73 who used the method of kriging, based on Gaussian processes, to search for systems with high melting point based on observed data. The same technique was recently applied to identify low thermal conductivity materials. Reference Seko, Togo, Hayashi, Tsuda, Chaput and Tanaka118 A particularly fruitful field of study in the future may be in the combination of such recommender systems with high-throughput computation. In such a scheme, the results of an initial set of computations would produce the initial data set needed for a recommender system to make further suggestions. These new suggestions would be computed automatically and then fed back into the recommender, thereby creating a closed loop that continually produces interesting candidates for follow up.
IV. APPLICATION EXAMPLE: VALENCE AND CONDUCTION BAND CHARACTER
The combination of open materials databases, programmatic APIs, and data mining techniques has recently exposed new and unprecedented opportunities to apply materials informatics. We next demonstrate work that analyzes the character of valence and conduction bands in materials over a large data set. This study was performed using only free software tools: the Materials Project database, Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 the MAPI programmatic API, Reference Ong, Cholia, Jain, Brafman, Gunter, Ceder and Persson59 the pymatgen code, Reference Ong, Richards, Jain, Hautier, Kocher, Cholia, Gunter, Chevrier, Persson and Ceder53 and the Python and R programming languages (including the BradleyTerry2 package Reference Turner and Firth119 implemented in R).
The aim of our study is to develop a probabilistic model for predicting the dominant character (element, charge state, and orbital type) of the electronic states near the valence band maximum (VBM) and conduction band minimum (CBM) over a broad range of compounds. Many materials properties, including the band gap (important for light capture) and Seebeck coefficient (important for thermoelectrics) depend critically on the details of these band edges. Understanding the character of these states allows one to determine the type of modifications needed to achieve desired properties.
Our strategy for developing a probabilistic model of VBM and CBM character can be outlined as follows:
-
(i) Select a computational data set from the Materials Project database and download the projected DOS information using the MAPI. The projected DOS contains the relative contributions of all the elements and orbitals in the material to the DOS at each energy level (see Fig. 9).
-
(ii) For each material, assign valence states to each element using a bond valence method; remove materials with mixed or undetermined valence.
-
(iii) Assess the contribution of each combination of element, valence, and orbital (e.g., O2−:p, hereafter called an ionic orbital) to the VBM and CBM, thereby assigning a score to each ionic orbital (based on Fermi weighting, see Supplementary Info) toward the VBM and CBM in that material.
-
(iv) For each material, and separately for the VBM and CBM, determine the higher score amongst all pairs of ionic orbitals. This can be considered a set of “competitions” within that material for greater contribution to the VBM and CBM for which there are binary win/loss records between pairs of ionic orbitals (e.g., O2−:p contributes more to the VBM than F1−:p). When iterated over all materials in the data set, this will result in a comprehensive set of pairwise rankings regarding which ionic oribtals tend to dominate the VBM/CBM.
-
(v) Use a Bradley–Terry model Reference Bradley and Terry120 to transform pairwise assessments of ionic orbitals into a single probabilistic ranking of which ionic orbitals are most likely to form the VBM and CBM states. This technique is useful because each material provides only a limited view (i.e., comparisons between the few elements contained in that material) of the overall ranking between all ionic orbitals. For example, this technique is one way to determine an absolute ranking of sports teams based only on pairwise competition data.

FIG. 9. Example of projected DOS used in the study (MoO3, entry mp-18856 in the Materials Project Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 ). The states near the valence band are dominated by O2−:p, whereas those near the conduction band are dominated by Mo6+:d. Over 2500 such materials are used to assess statistics on valence and conduction band character in this study.
The details of our methodology, including considerations such as the treatment of d orbitals in the Materials Project dataset, are presented in the Supplementary Information.
The results of our study for a selected set of ionic orbitals are presented in Fig. 10; the full results are presented in the Supplementary Information. Each data point in Fig. 10 represents the probability that the ionic orbital listed on the y axis will have a greater contribution to the specified band edge than the orbital listed on the x axis. The ionic orbitals that tend to dominate are ordered top-to-bottom along the y axis, and left-to-right along the x axis. For example, the data indicate that in a material containing both Cu1+:d and Fe3+:d states, the VBM is highly likely to be dominated by Cu1+:d (probability close to 1.0) where as the CBM is more likely to be dominated by Fe3+:d. We note that our model should not be interpreted as a ranking of energy levels in compounds. For example, an ionic orbital may have a low VBM score either because the energy tends to be too high (it forms a conduction band) or too low (too deep in the valence band).

FIG. 10. Pairwise probability for the ionic orbital listed to the left to have a greater contribution to a band edge then the ionic orbital listed at the bottom. The VBM edge data are listed to the left, and the CBM data to the right. Only a selected set of ionic orbitals are depicted; the full data set is in the Supplementary Information.
As an example of how this type of analysis can be used in materials science problems, the absolute VBM position of a material has been demonstrated to be correlated with its ease to be p-type. Reference Robertson and Clark121–Reference Zunger123 Oxides with purely oxygen valence band character tend to have a too low valence band to be p-type dopable. The design principle of using cations that will hybridize with oxygen to lift the valence band up and facilitate p-type doping is an important paradigm in the development of new p-type oxides and especially transparent conducting oxides (TCOs). The presence in our data set of Cu1+ or Ag1+ as cations that compete strongly with oxygen is consistent with the use of these elements in p-type TCOs. Reference Kawazoe, Yasukawa and Hyodo124–Reference Hautier, Miglio, Ceder, Rignanese and Gonze129 Notably, other 3d ions in our analysis, including V3+ and Mn2+, are also known to form p-type oxides. Reference Peng and Lany130 This analysis can therefore be used as a first step toward the more systematic listing of ions necessary to form p-type oxides.
In some cases, the data do not match our intuition; for example, we expect that S2−:p would be ranked higher in the valence band than O2−:p. Counter to our result that O2−:p and S2−:p are ranked similarly, in all specific examples in our data set for which a single material simultaneously contained both O2−:p and S2−:p orbitals, the greater VBM contribution came from S2−:p. This discrepancy suggests that a consistent universal ranking of ionic orbitals may not exist. For example, when considering compounds containing Mn2+:d, the VBM is composed mostly of S2−:p in only 20% of cases versus 88% for O2−:p (see the Supplementary Info). An example of the latter is MnPO4 with spacegroup Pmnb (mp-777460 on the Materials Project Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson38 web site), in which O2−:p dominates the VBM, whereas Mn2+:d forms the CBM. Thus, interactions between ionic orbitals can be in conflict depending on a specific material's physics. Possible improvements to the model may include more heavily weighting direct competitions, or taking into account the relative energies between ionic orbitals. Nevertheless, overall our scores are in good agreement with known principles and provide general guidelines for engineering band edges.
Our model for predicting VBM and CBM characters is one example of how it is now possible to leverage open materials data sets and software tools to provide rapid and data-driven models for problems normally guided by intuition and experience. However, it should be noted that the results of such approaches are still limited by the accuracy of the underlying data set. In the case of this analysis, the generalized gradient approximation with +U correction (GGA+U) method was used by the Materials Project to handle localized/correlated d states, but this method is not universally accurate. Nevertheless, the fact that today, these models can be built on top of already existing data sets and software libraries speaks to the potential democratic power of materials informatics.
V. DISCUSSION AND CONCLUSION
Data mining and informatics-based approaches present new opportunities for materials design and understanding. As the amount of publicly available materials data grows, such techniques will be able to extract from these data sets scientific principles and design rules that could not be determined through conventional analyses. In this paper, we reviewed some of these emerging materials databases along with several techniques and examples of how materials informatics can contribute to materials discovery. We also demonstrated how one can already combine open data and software tools to produce an analysis predicting the character of a compound's valence and conduction bands. However, despite these opportunities, several challenges remain that have limited the impact of materials informatics approaches thus far and will no doubt be the subject of future work.
The first remaining challenge is for the community to gain experience in navigating the challenges of using large data resources. Even as the number of databases grows and as programmatic APIs to the data become available, the fraction of users that extract and work with large data sets remains relatively small. Furthermore, when the desired data are not available from a single source, it is difficult to query over multiple resources and to combine information from different databases. It is especially challenging to match computations against experimental measurements because data regarding the crystal structure and other conditions under which the experiments were performed is usually missing. Working with computational data alone is also tricky; researchers must understand the error bars of the simulation method, which can sometimes be quite high and difficult even for expert users to estimate. Thus, even though the situation for materials data is improving rapidly, today one must still be somewhat of an expert user to exploit these resources.
The second challenge is the development of materials descriptors for crystalline, periodic solids. While there has been progress in the last several years, this area is still ripe for new ideas. Today, we still do not possess automatic algorithms that can describe crystals using descriptors that would typically be used by a domain expert. Such descriptors could include the nature and connectivity of local environments (e.g., “edge-sharing tetrahedra” versus “corner-sharing octahedra”), qualitative assessments of structure (e.g., “close-packed” versus “layered” versus “1D channels”), or crystal prototype (e.g., “double perovskite” or “ordering of rock salt lattice”). Crystal structure data are quite complex and varied. Without such algorithms, it is difficult for researchers to describe a crystal to a machine learning algorithm using constructs proven to be successful by decades of materials science.
Finally, the third challenge is in assessing the appropriateness and transferability of machine learning models. Typically, such assessments are made using a performance-oriented metric such as cross-validation error. However, there are many reasons why such performance metrics might be misleading. The first issue is that the cross-validation error can be affected by the type of cross-validation (e.g., leave-one-out versus n-fold) Reference Arlot and Celisse131 as well as the design of how models are selected and how the data are split for fitting (i.e., training, validation, and test sets). Reference Bell, Koren and Volinsky106 However, perhaps an even bigger concern is in controlling and reporting sampling errors, i.e., in training and cross-validating a model on a sample that is not representative of the full population. Examples of potential sampling error include (i) building and validating a model on a data set of binary compounds and subsequently applying that error estimation to ternary and quaternary compounds, (ii) building and validating a model's performance for a limited number of highly symmetric structure types, and then applying the same model to predict the behavior of dissimilar crystal structures, and (iii) training the model only on thermodynamically stable compounds and then applying that model to unstable compounds. In many cases, it is very difficult to obtain data for samples that are truly representative of the prediction space, and better methods to estimate error and applicability are needed for these situations.
This third issue of knowing how much weight to put in a machine learning model is particularly important because the models that achieve the lowest cross-validation error are often the most complex (e.g., neural nets or random forests) and can be impossible to interpret scientifically. Should materials scientists trust a prediction from an impenetrable machine learning model when it is in conflict with the intuition afforded by a more classical, interpretable model? Part of the distinction lies in whether the goal of machine learning models is to simply be predictive (capable of making useful forecasts) or whether additional weight should be afforded to models that are causal (confirming that a factor is truly the reason behind an effect) or mechanistic (reproduce the physics behind an effect). Ideally, machine learning models will not only try to answer specific questions accurately but will also prove useful in leading us to better questions and to new types of analyses (as was the case for the periodic table).
While it will take some time to truly develop solutions to all of these challenges, informatics is already making headway into materials science, and data-driven methods will no doubt form a major area in the study of materials in the future.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1557/jmr.2016.80.
ACKNOWLEDGMENTS
This work was intellectually led by the Materials Project (DOE Basic Energy Sciences Grant No. EDCBEE). Work at the Lawrence Berkeley National Laboratory was supported by the U.S. Department of Energy Office of Science, Office of Basic Energy Sciences Department under Contract No. DE-AC02-05CH11231. GH acknowledges financial support from the European Union Marie Curie Career Integration (CIG) grant HT4TCOs PCIG11-GA-2012-321988. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility.












