



Introduction
Phase-change materials (PCMs) are employed in storage devices (rewritable optical discs, non-volatile electronic memories), which exploit their ability to undergo fast and reversible transitions between the crystalline and amorphous state.[Reference Wuttig and Yamada1–Reference Zhang, Deringer, Dronskowski, Mazzarello, Ma and Wuttig3] Long-term information storage is possible, thanks to the optical and electronic contrast between the two phases, as well as the high stability of the amorphous state near room temperature. Phase-change storage-class memories have recently entered the market[4, Reference Choe5]: these devices are significantly faster than flash memories, and thus, they fill the gap between DRAM and solid-state drives in terms of performance. The most important family of PCMs for memory applications belongs to the pseudobinary line connecting GeTe with Sb2Te3.[Reference Wuttig and Yamada1–Reference Zhang, Deringer, Dronskowski, Mazzarello, Ma and Wuttig3, Reference Rao, Ding, Zhou, Zheng, Xia, Lv, Song, Feng, Ronneberger, Mazzarello, Zhang and Ma6–Reference Xie, Kim, Kim, Kim, Gonsalves, BrightSky, Lam, Zhu and Cha11] In particular, Ge2Sb2Te5 (GST) is currently used in electronic memories. Crystallization in PCMs is slower than the reverse amorphization process; hence, the former determines the maximum speed of the device. Recently, prototype phase-change cells containing Sc-alloyed Sb2Te3 (Sc0.2Sb2Te3) have shown subnanosecond crystallization.[Reference Rao, Ding, Zhou, Zheng, Xia, Lv, Song, Feng, Ronneberger, Mazzarello, Zhang and Ma6] Such switching speed is comparable with that of SRAM, and could lead to the development of a universal phase-change memory.
Considerable experimental and theoretical work has been devoted to the study of the strong temperature dependence of the dynamics of PCMs.[Reference Orava, Greer, Gholipour, Hewak and Smith12–Reference Orava, Weber, Kaban and Greer18] This is a key property that ensures said fast crystallization at elevated temperatures and high stability of the amorphous phase at low temperatures. Such property has been linked to the high fragility of the supercooled liquid state, namely the fact that the viscosity behaves in a non-Arrhenius fashion.[Reference Orava, Greer, Gholipour, Hewak and Smith12] The atomistic processes involved in the high-temperature crystallization of PCMs have also been addressed by several computational studies based on density–functional–theory (DFT) molecular dynamics (the so-called ab initio molecular dynamics––AIMD) or accurate neural-network potentials fitted to DFT data.[Reference Hegedüs and Elliott19–Reference Zhang and Ma33]
Upon fast crystallization of the glass, GST forms a metastable rocksalt phase,[Reference Raty, Bichara, Mazzarello, Rausch, Zalden and Wuttig34] in which Te atoms occupy one sublattice, while Ge, Sb, and vacancies occupy the second one in a random fashion. It has been shown experimentally[Reference Siegrist, Jost, Volker, Woda, Merkelbach, Schlockermann and Wuttig35–Reference Bragaglia, Arciprete, Zhang, Mio, Zallo, Perumal, Giussani, Cecchi, Boschker, Riechert, Privitera, Rimini, Mazzarello and Calarco37] and theoretically[Reference Zhang, Thiess, Zalden, Zeller, Dederichs, Raty, Wuttig, Blügel and Mazzarello38–Reference Xu, Zhang, Mazzarello and Wuttig40] that the degree of disorder in rocksalt GST and similar compounds on the pseudobinary line can be tuned by thermal annealing or even pressure, so as to induce Anderson insulator–metal transitions induced by disorder.
GST is a nucleation-dominated PCM, implying that crystallization of an amorphous mark occurs via formation and subsequent growth of crystalline nuclei. AIMD simulations of these processes are challenging, due to the small time- and length-scales accessible and the stochastic nature of nucleation.
In our previous work,[Reference Ronneberger, Zhang, Eshet and Mazzarello26] we considered models of GST containing about 500 atoms and performed two sets of simulations. In one set, we generated amorphous models inside a crystalline matrix (obtained by keeping two crystalline layers fixed during melting and quenching), and studied crystal growth at the planar crystal–amorphous interface. In the second one, we employed the metadynamics method[Reference Laio and Parrinello41] (a powerful and versatile enhanced-sampling technique) to accelerate the formation of postcritical nuclei and then monitored the growth of these nuclei during unbiased AIMD simulations. Here, we carry out large-scale AIMD simulations of crystallization of 900-atom models of GST at high temperatures ranging between 600 and 800 K. Similarly to our previous work, the crystallization kinetics is determined from a crystalline nucleus, which is created by means of metadynamics. The goal of this work is twofold: (a) address finite-size effects by comparing these simulations with previous work employing smaller models; (b) determine how temperature affects crystal growth and the size of the critical nucleus.
Methods
We perform large-scale AIMD simulations based on the “second-generation” Car–Parrinello scheme,[Reference Kühne, Krack, Mohamed and Parrinello42, Reference Kühne43] which is implemented in the Quickstep code of the CP2K package.[Reference Hutter, Iannuzzi, Schiffmann and VandeVondele44] The generalized gradient approximation to the exchange-correlation potential[Reference Perdew, Burke and Ernzerhof45] and the scalar-relativistic Goedecker pseudopotentials[Reference Goedecker, Teter and Hutter46] are employed. A triple-zeta plus polarization Gaussian-type basis set is used to expand the Kohn–Sham orbitals, whereas plane waves with a cutoff of 300 Ry are used to expand the charge density. The Brillouin zone is sampled at the Γ point of the supercell and the periodic boundary conditions are imposed. The time step is 2 fs. The temperature is controlled by a stochastic Langevin thermostat. The simulations are conducted in the canonical ensemble (NVT) with a fixed density of 0.03173 Å lying between the experimental amorphous value (0.0300 Å) and the crystalline one (0.03283 Å). Fixing the density during the simulation is relevant to phase-change memory cells, where the presence of a crystalline matrix constrains the volume available during recrystallization of the amorphous mark. A purely amorphous model containing 900 atoms in a cubic 3.11 nm × 3.11 nm × 3.11 nm supercell is obtained by a melt-quenching procedure. In this procedure, the model is equilibrated at the melting temperature for 30 ps and then quenched to the room temperature with a quenching rate of 1013 K/s. Subsequently, a crystalline nucleus inside the amorphous model is created by running a metadynamics simulation. Metadynamics[Reference Laio and Parrinello41] exploits the idea of a bias potential acting on a small set of collective variables (CVs) that describe the relevant degrees of freedom of the system. It allows one to efficiently explore the phase space and to estimate the free energy changes in a molecular dynamics simulation.[Reference Laio and Parrinello41, Reference Quigley and Rodger47] The CVs ξ i, i = 1, … , d are functions of the atomic coordinates and should be able to characterize the process of interest. During the MD run, a history-dependent bias potential v g is added to the potential energy of the system. The bias potential is given by a sum of Gaussians in the d-dimensional space of the CVs. The Gaussians are centered at values 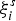 $\xi _i^s $ taken at the time intervals τ, 2τ, 3τ, … , < t:
$\xi _i^s $ taken at the time intervals τ, 2τ, 3τ, … , < t:
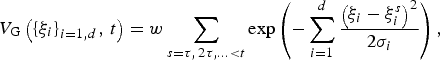 $$V_{\rm G}\left( {{\left\{ {\xi _i} \right\}}_{i = 1,d},\,t} \right) = w\mathop \sum \limits_{s = \tau, \,2\tau, \ldots \lt t} \exp \left( { - \mathop \sum \limits_{i = 1}^d \displaystyle{{{\left( {\xi _i - \xi _i^s} \right)}^2} \over {2\sigma _i}}} \right),$$
$$V_{\rm G}\left( {{\left\{ {\xi _i} \right\}}_{i = 1,d},\,t} \right) = w\mathop \sum \limits_{s = \tau, \,2\tau, \ldots \lt t} \exp \left( { - \mathop \sum \limits_{i = 1}^d \displaystyle{{{\left( {\xi _i - \xi _i^s} \right)}^2} \over {2\sigma _i}}} \right),$$where w and σ i denote the height and width of the Gaussians and control the accuracy and efficiency of the simulation.
Results
In standard AIMD runs, the generation of a crystalline nucleus is computationally extremely heavy, since nucleation is a stochastic process. The first computational studies of crystallization kinetics of GST used very small cells containing <200 atoms,[Reference Hegedüs and Elliott19, Reference Lee and Elliott20] which facilitated crystallization. Subsequent studies on larger systems were either focused on models containing a crystalline seed, which was fixed during the melt-quench procedure,[Reference Kalikka, Akola, Larrucea and Jones21] or were based on very long simulation times of the order of a few nanoseconds to enable the formation of large nuclei.[Reference Kalikka, Akola and Jones24, Reference Kalikka, Akola and Jones27] In Ref. Reference Ronneberger, Zhang, Eshet and Mazzarello26, we employed metadynamics to create a postcritical crystalline nucleus inside a 460-atom model at T = 600 K. In this study, we follow a similar procedure and run a metadynamics simulation prior to the unbiased run of crystallization. Since we are only interested in accelerating the nucleation and do not aim at the estimation of free energies, we sample the phase space in a coarse and efficient manner using large Gaussians (w = 0.5 meV/atom, σ 1 = 0.01, σ 2 = 3 meV/atom, τ = 0.1 ps) and employing the multiple walkers scheme[Reference Raiteri, Laio, Gervasio, Micheletti and Parrinello48] with seven independent walkers. In the multiple walker scheme, the sampling speed is increased by simultaneously running independent simulations that add Gaussians to the same shared bias potential. The metadynamics run is conducted at ~600 K, which is also one of the target temperatures of the unbiased simulations. The most important step for the success of a metadynamics simulation is the choice of appropriate CVs. In the case of nucleation, they should be able to discern atoms in a crystalline-like environment. One of the CVs in our metadynamics simulation is based on the local parameter 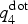 $q_4^{{\rm dot}} $,[Reference ten Wolde, Ruiz-Montero and Frenkel49] which is computed for each atom and measures the average correlation of the bonding arrangement between the atom and its neighboring atoms. It takes values close to one for atoms in a crystalline-like environment and close to zero for atoms in a disordered, amorphous-like environment. The top left panel of Fig. 1 shows the distributions of
$q_4^{{\rm dot}} $,[Reference ten Wolde, Ruiz-Montero and Frenkel49] which is computed for each atom and measures the average correlation of the bonding arrangement between the atom and its neighboring atoms. It takes values close to one for atoms in a crystalline-like environment and close to zero for atoms in a disordered, amorphous-like environment. The top left panel of Fig. 1 shows the distributions of 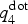 $q_4^{{\rm dot}} $ in the amorphous and the crystalline phases: since the two distributions display almost no overlap, this quantity indeed allows to discriminate the two phases and is therefore well suited as a CV in nucleation studies. By setting a threshold value of 0.45, we define an atom to be crystalline-like if its
$q_4^{{\rm dot}} $ in the amorphous and the crystalline phases: since the two distributions display almost no overlap, this quantity indeed allows to discriminate the two phases and is therefore well suited as a CV in nucleation studies. By setting a threshold value of 0.45, we define an atom to be crystalline-like if its 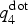 $q_4^{{\rm dot}} $ value exceeds this threshold. This definition is also used for the analysis of the crystal growth. The CV in our metadynamics simulation is obtained by averaging
$q_4^{{\rm dot}} $ value exceeds this threshold. This definition is also used for the analysis of the crystal growth. The CV in our metadynamics simulation is obtained by averaging 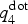 $q_4^{{\rm dot}} $ inside a spherical region around a preselected atom. The size of the spherical region is chosen to be large enough to encompass a postcritical nucleus (which, in our previous work, was estimated to contain <150 atoms at 600 K), but as small as possible to minimize the finite size effects due to the periodic boundary conditions. As second CV, we use the potential energy of the system U.
$q_4^{{\rm dot}} $ inside a spherical region around a preselected atom. The size of the spherical region is chosen to be large enough to encompass a postcritical nucleus (which, in our previous work, was estimated to contain <150 atoms at 600 K), but as small as possible to minimize the finite size effects due to the periodic boundary conditions. As second CV, we use the potential energy of the system U.
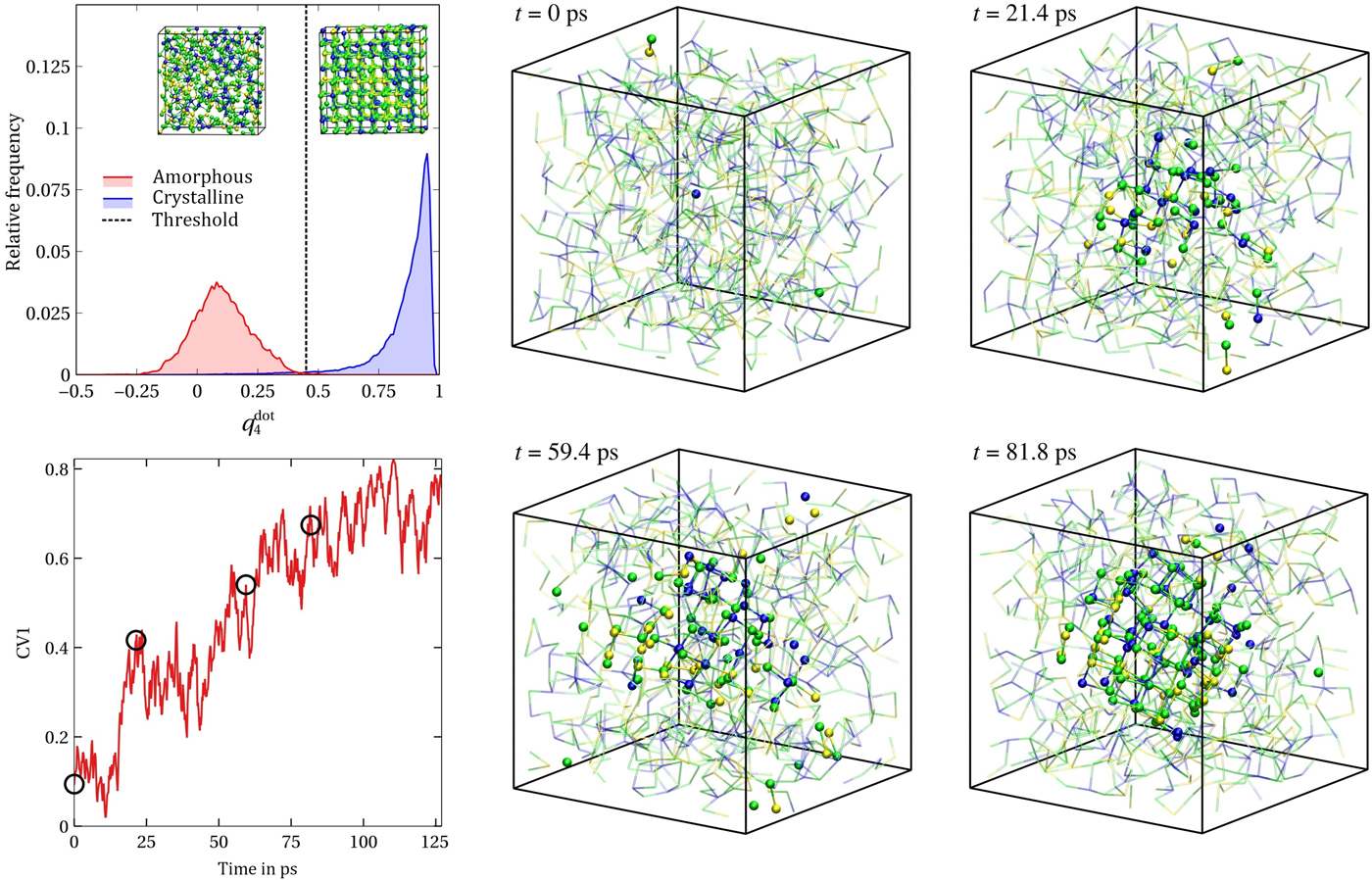
Figure 1. (Left) Distribution of 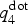 $q_4^{{\rm dot}} $ in the amorphous and the crystalline phases (top) and evolution of the averaged
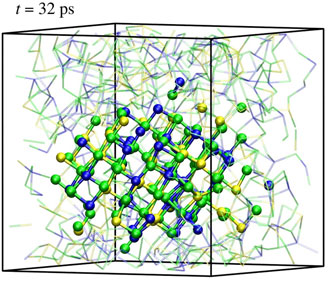
$q_4^{{\rm dot}} $ in the amorphous and the crystalline phases (top) and evolution of the averaged 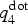 $q_4^{{\rm dot}} $ employed in the metadynamics simulation as first CV (see also main text) for a selected walker. The insets in the top panel show the typical atomic structures of amorphous and cubic rocksalt GST. (Right) Formation of a crystalline nucleus of GST (solid spheres and sticks) inside the amorphous phase (small, transparent sticks and spheres). Te atoms are green, Sb atoms are yellow, and Ge atoms are blue. The snapshots are taken at the corresponding time steps (black circles) of the biased simulations shown in the bottom left plot of the CV.
$q_4^{{\rm dot}} $ employed in the metadynamics simulation as first CV (see also main text) for a selected walker. The insets in the top panel show the typical atomic structures of amorphous and cubic rocksalt GST. (Right) Formation of a crystalline nucleus of GST (solid spheres and sticks) inside the amorphous phase (small, transparent sticks and spheres). Te atoms are green, Sb atoms are yellow, and Ge atoms are blue. The snapshots are taken at the corresponding time steps (black circles) of the biased simulations shown in the bottom left plot of the CV.
As a result of the metadynamics simulations, some of the walkers sample the relevant region of the CV space where a crystalline nucleus starts to form. The bottom left panel of Fig. 1 shows the time evolution of the first CV for one such walker. On the right panel of Fig. 1, the formation of a crystalline nucleus inside the amorphous phase is visualized at a few selected snapshots corresponding to the times shown as black circles in the left bottom panel. Although size and shape of the nucleus can vary significantly due to the fluctuations of the CVs, we obtain a single, compact crystalline cluster at final stages of this walker.
Subsequently, we performed a long, unbiased simulation at ~600 K starting from a snapshot with a sizable crystalline nucleus containing about N c ~175 crystalline-like atom. The crystallization process completed in ~810 ps and the recrystallized structure was in a metastable cubic phase. Three representative snapshots of the growing crystalline nucleus (solid balls) within the amorphous phase (transparent bonds) are depicted in Fig. 2. The crystalline nucleus is initially quasi-spherical; however, it becomes elongated after some transient period. The presence of a geometrically complex amorphous–crystal interface makes the computation of the crystal growth velocity v g difficult. Considering a general crystalline volume V c with a bounding surface S ac, which divides the crystalline and amorphous regions, we define the v g as the average flux through the surface:
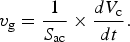 $$v_{\rm g} = \displaystyle{1 \over {S_{{\rm ac}}}} \times \displaystyle{{dV_{\rm c}} \over {dt}}.$$
$$v_{\rm g} = \displaystyle{1 \over {S_{{\rm ac}}}} \times \displaystyle{{dV_{\rm c}} \over {dt}}.$$
Here we use the Voronoi tessellation of the supercell in order to define V c and S ac. We obtain V c by summing up the volumes of the Voronoi polyhedra of each crystalline-like atom. S ac is the total area of the faces that are shared by Voronoi polyhedra of amorphous-like and crystalline-like atoms. We use the voro++ code[Reference Rycroft50] to compute the Voronoi polyhedra in our models. In Fig. 3, we compare the 900-atom model with a 460-atom model from our previous work[Reference Ronneberger, Zhang, Eshet and Mazzarello26] by plotting V c, S ac, and v g as a function of time. This allows us to assess the finite size effects on the estimation of v g. The time derivative dV/dt is calculated by smoothing V c with a Savitzky–Golay filter with a window size 2 ps. The computed velocities are only meaningful in the range in which V c is increasing. The start and end of this range are indicated by thin, vertical lines in Fig. 3. The crystallization of the larger model containing 900-atom requires considerably longer simulation time as compared with the 460-atom models, because of the larger amorphous volume and the lower crystal growth velocity. After a closer inspection of the v g curve for the 900-atom model, we can divide the crystallization of the 900-atom model into three distinct regimes: two regimes with a plateau of v g values around ~0.5 and ~0.8 m/s and a third one with a peak value around ~1.4 m/s. In the first two regimes, the amorphous–crystalline interface area S ac is increasing and the growth velocity is dominated by the derivative dV c/dt. In the first regime, the crystalline nucleus is small enough so that it is not connected with its periodic images or, at most, elongated along one direction. It corresponds to the crystalline nucleus shown in the left snapshot in Fig. 2. In this regime, the amount of amorphous volume is maximal and the finite size effects are minimal. Therefore, this region provides the best estimate for v g. In the second regime, the crystalline nucleus is connected with its periodic image, as in the middle snapshot in Fig. 2. The crystalline volume v c of the nucleus becomes roughly comparable to the volume of the smaller 460-atom model of Ref. Reference Ronneberger, Zhang, Eshet and Mazzarello26. The slope dV/dt of the 900-atom model in this regime is similar to that of the 460-atom at the initial stage, so that the values of v g fall into a similar range close to ~1 m/s. In the final regime, the surface effects dominate, since 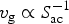 $v_{\rm g} \propto S_{{\rm ac}}^{ - 1} $ and there is an abrupt decrease in S ac due to the vanishing amorphous region in the last stages of the crystallization process. Overall, we conclude that the finite size effects in the 460-atom models lead to a non-negligible overestimation of v g and its values are roughly doubled by going from 900 to 460 atoms.
$v_{\rm g} \propto S_{{\rm ac}}^{ - 1} $ and there is an abrupt decrease in S ac due to the vanishing amorphous region in the last stages of the crystallization process. Overall, we conclude that the finite size effects in the 460-atom models lead to a non-negligible overestimation of v g and its values are roughly doubled by going from 900 to 460 atoms.
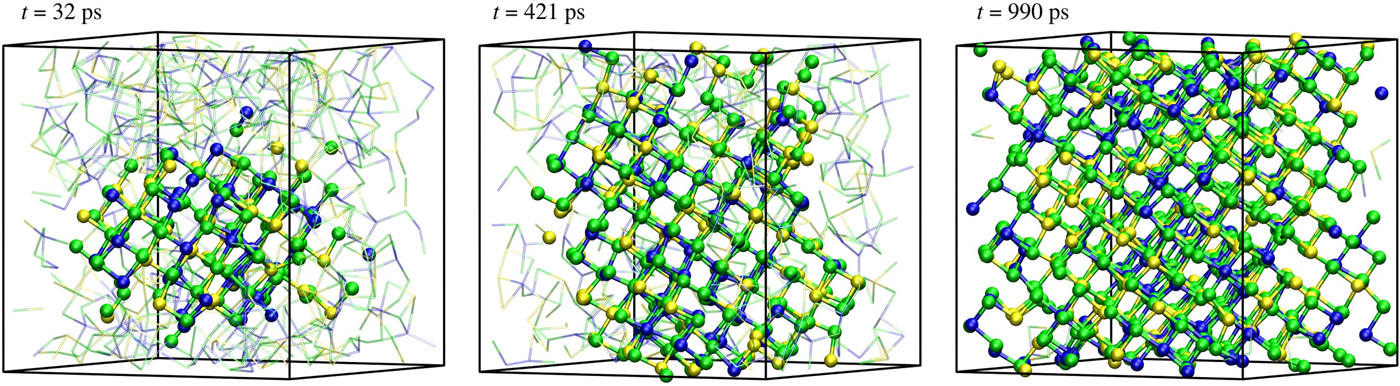
Figure 2. Unbiased simulations at ~600 K. Rapid crystal growth around the crystalline nucleus generated by metadynamics are observed. The snapshots are taken at t = 32, 421, 990 ps of the unbiased simulations.
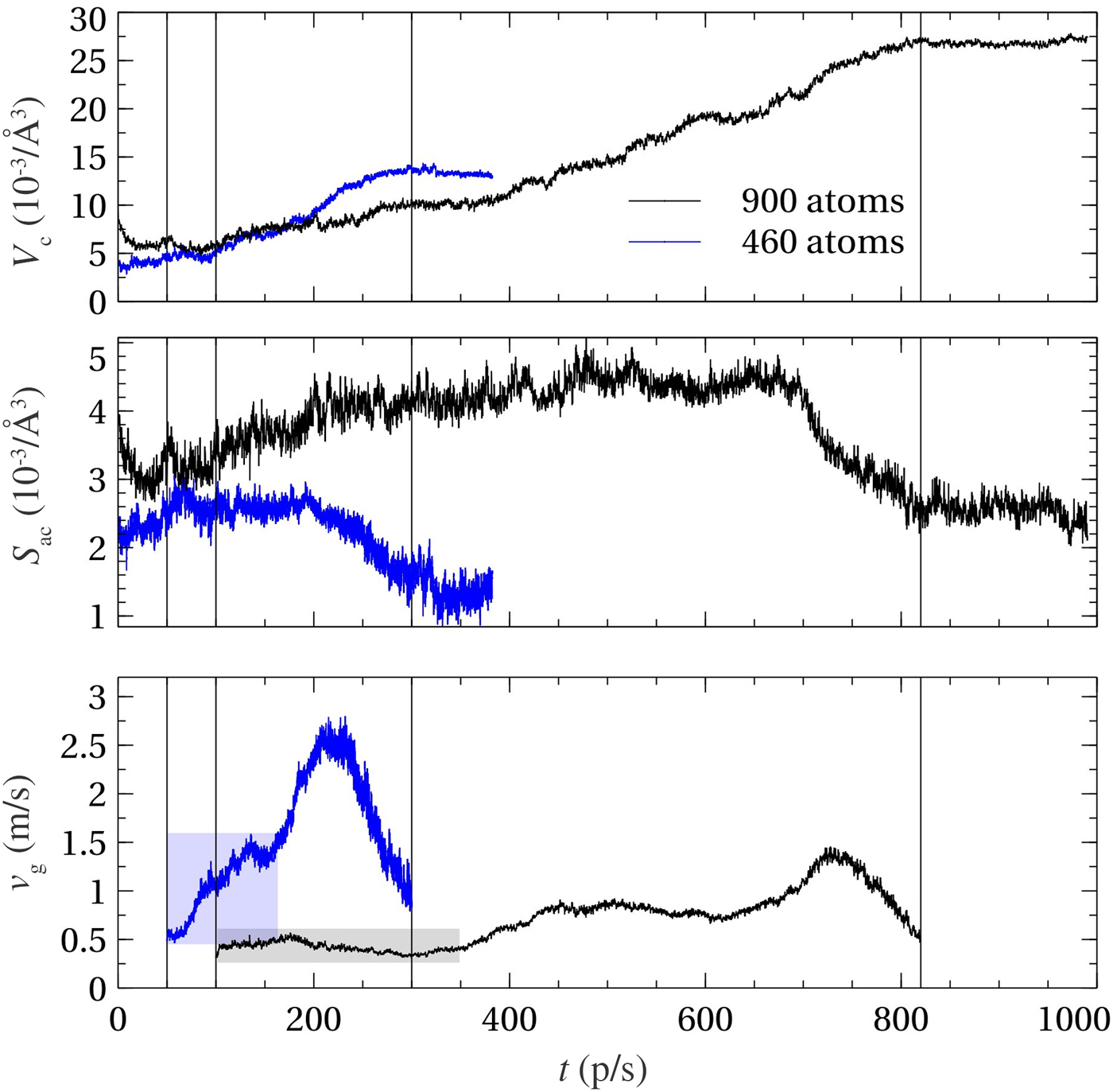
Figure 3. Evolution of the crystalline volume V c (top), amorphous–crystal interface area S ac (middle) and the corresponding crystal growth velocity (bottom) for 900- and 460-atom models of GST at ~600 K. Vertical lines indicate the start and end points of the time window in which V c is increasing.
Next we address the crystal growth velocities at higher temperatures. In principle, independent amorphous models containing a postcritical crystalline nucleus at the target temperature should be considered to accurately estimate v g. Since such simulations would be computationally too demanding, we consider a simpler approach that allows us to approximately assess the crystallization kinetics at high temperatures. It has to be noticed, however, that the higher the temperature, the stronger the finite-size effects (for a fixed simulation box), since the size of the critical nucleus typically increases with temperature. The latter property stems from the fact that the interfacial energy is weakly dependent on temperature, whereas the chemical potential difference between the two phases (which, according to classical nucleation, is inversely proportional to the radius of the critical nucleus[Reference Orava and Greer51]) decreases roughly linearly with temperature. Concretely, we perform a new set of simulations at ~700 and ~800 K starting from a snapshot at t = 222 ps of the crystallizing model at ~600 K. The choice of the starting configuration at a later stage is appropriate, because the size of the critical nucleus is larger at higher temperatures, as mentioned above. In our case, the crystalline nucleus already contains 245 atoms in this starting configuration. The simulation temperature is raised directly from ~600 K to the target temperatures. In Fig. 4, we show the evolution of the crystalline volumes V c for the three sets of simulations (at ~600, ~700, and ~800 K, respectively) and the corresponding crystal growth velocities. At ~700 K, the crystalline cluster starts to grow after a short stabilization period. This indicates that its size is large enough to be postcritical. By contrast, the nucleus is not sufficiently large to grow at ~800 K. Instead, the cluster shrinks and dissolves quickly at this temperature. Therefore, we only plot v g for the simulations at ~600 and ~700 K in Fig. 4. The initial crystallization regimes with minimal finite size effects are indicated with shaded rectangular regions. From this, we extract a crystal growth velocity v g~1.5 m/s at ~700 K, which is approximately three times larger than the one at ~600 K. This trend is in line with kinetic measurements based on ultrafast differential scanning calorimetry,[Reference Orava, Greer, Gholipour, Hewak and Smith12] where the maximum growth rate is reached at temperatures close to 700 K.
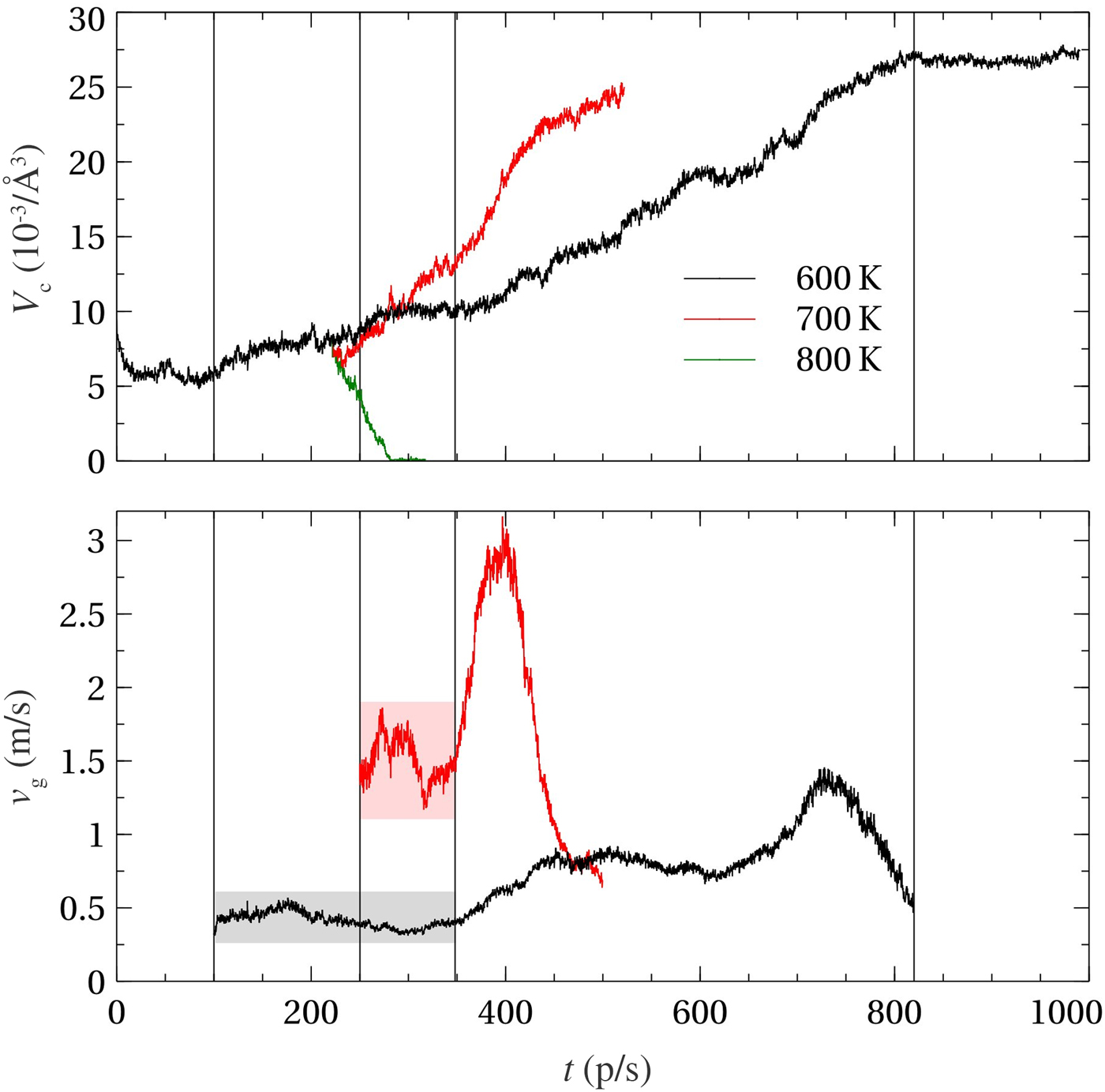
Figure 4. Evolution of the crystalline volume V c (top) and the corresponding crystal growth velocity (bottom). The model is heated up to ~700 and ~800 K after running for 222 ps at ~600 K.
Conclusions
In this work, we have studied the crystallization kinetics of a large model of GST containing 900 atoms starting from a crystalline nucleus of about 175 atoms. The crystalline nucleus was successfully generated from a metadynamics simulation with an appropriate set of CVs. Comparing the crystal growth velocities for this model with the ones obtained for a smaller 460-atom model in our previous work, we have assessed the finite size effects and found a reduced crystal growth velocity of ~0.5 m/s in the larger model, which is roughly half of the value obtained for the 460-atom model. The crystal growth velocity at a higher temperature of ~700 K is estimated to be v g~1.5 m/s from a single run with an initial configuration containing a crystalline cluster of 245 atoms. At even higher temperature of ~800 K, the latter crystalline nucleus is found to be unstable.
Acknowledgments
The authors acknowledge the computational resources granted by JARA-HPC from RWTH Aachen University under project JARA0150, as well as funding by the DFG (German Science Foundation) within the collaborative research centre SFB 917 “Nanoswitches.” W.Z. gratefully thanks the support of the National Natural Science Foundation of China (61774123), the Science and Technology Department of Jiangsu Province under the grant agreement No. BK20170414, the Youth Thousand Talents Program of China and the Young Talent Support Plan of Xi'an Jiaotong University. The support by the International Joint Laboratory for Micro/Nano Manufacturing and Measurement Technologies (IJL-MMMT) of Xi'an Jiaotong University is also acknowledged.





 Open access
Open access