



Introduction
Mites are incredibly diverse and abundant in the world, with an estimated total species count between 500 000 and 1 000 000, although only around 55 000 species have been described (Manu et al., Reference Manu, Băncilă, Bîrsan, Mountford and Onete2021). Mites have a complex habitat and a wide range of feeding activities, acting as predators, phytophagous, decomposers and fungivores to scavengers or parasitic on other animals (Badieritakis et al., Reference Badieritakis, Fantinou and Emmanouel2014). Currently, some Mesostigmata species have been shown to be potential vectors of zoonotic diseases and, together with small rodents, play an important role in the distribution of viruses (hantavirus, Saint-Louis encephalitis virus, tick-borne encephalitis virus, etc.), bacteria (Francisella tularensis, Salmonella typbimurium, Borrelia bugdorferi, etc.) and protozoan infections (Trypanosoma cruzi, etc.) (Moro et al., Reference Moro, Chauve and Zenner2005).
The genus Eulaelaps is zoologically classified under Acari, Parasitiformes, Mesostigmata, Gamasina and Haemogamasidae (Vinarski and Korallo-Vinarskaya, Reference Vinarski and Korallo-Vinarskaya2017). Most species of the genus Eulaelaps are blood-sucking parasites, found mainly in the nests and shelters of small mammals (Vinarski and Korallo-Vinarskaya, Reference Vinarski and Korallo-Vinarskaya2017). Eulaelaps stabularis is considered to be the most common mite found on the nests and bodies of small rodents and insectivores (Turk, Reference Turk1945; Allred, Reference Allred1969). Some mites of the genus Eulaelaps have morphological characters that are very similar, leading to a high degree of confusion in the identification of mites of the genus Eulaelaps by different scholars. For example, synonyms of E. stabularis include Gamasus stabularis, E. arcualis, E. oribatoides, E. oudemansi, Laelaps propheticus, etc. (Korneev, Reference Korneev2003; Vinarski and Korallo-Vinarskaya, Reference Vinarski and Korallo-Vinarskaya2017). Synonyms of E. kolpakovae include E. novus (Vinarski and Korallo-Vinarskaya, Reference Vinarski and Korallo-Vinarskaya2017). Some mites of the genus Eulaelaps carry a variety of zoonotic pathogens, e.g., E. stabularis and E. shanghaiensis can transmit hemorrhagic fever with renal syndrome (Moro et al., Reference Moro, Chauve and Zenner2005; Huang et al., Reference Huang, Guo, Speakman and Dong2013), and E. kolpakovae carries Yersinia pestis (Sludsky, Reference Sludsky2014).
The mitochondria are a semi-autonomous organelle found in almost all eukaryotes and are the main site of oxidative phosphorylation (Bi et al., Reference Bi, Lu, Xu, He and Lu2020). The mitochondrial genome, with its simple structure, matrilineal inheritance, low recombination rate, and rapid evolution, has been widely used in studies of parasite classification, species identification, molecular evolution, phylogenetics and population genetics (Gao et al., Reference Gao, Zhang, Wang and Zhao2022; Yang et al., Reference Yang, Yang, Chen and Dong2022). The mitochondrial genome is usually composed of 37 genes, that is, 13 protein-coding genes (PCGs), 22 transporter RNA genes (tRNAs), 2 ribosomal RNA genes (rRNAs) and a non-coding region (control regions, CR) (Yang et al., Reference Yang, Chen and Dong2023). In recent years, with the rapid development of genome assembly technologies and the gradual reduction of sequencing costs, the mitochondrial genomes of an increasing number of species have been sequenced. At the same time, the use of mitochondrial genomes to explore the phylogenetic relationships among species has become a hot topic of research (Cameron, Reference Cameron2014; Chen et al., Reference Chen, Chen, Liao, Wang and Wang2022b).
Currently, for none of the species within the genus Eulaelaps mitochondrial genomes have been sequenced. In order to further fill the gap in understanding the molecular evolution of species within the genus Eulaelaps. Hence, in this study, the mitochondrial genome of E. silvestris parasitizing Apodemus chevrieri was sequenced and assembled, and a phylogenetic tree was constructed using molecular systematics-related methods. The results provide a valuable resource for further research on the genetic diversity and phylogenetic analysis of mites in the genus Eulaelaps.
Materials and methods
Sample collection and morphological identification
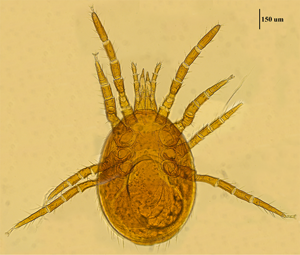
Mite specimens were collected from the body surface of A. chevrieri in Hongyuan County, Sichuan Province, China. Collected mite specimens were stored in EP tubes containing 95% ethanol, and the sample number was labeled 161. In the laboratory, collected mite specimens were directly placed under a SZ2-ILST dissecting microscope (Olympus, Tokyo, Japan) for species identification. One sample was taken and mounted on a glass slide with Hoyer's solution, and after dehydration, drying and transparency, the mounted sample was photographed under a Leica DM 3000 LED microscope (Lecia, Weztlar, Germany) to obtain pictures of the morphological characteristics of the sample. The mite was morphologically identified according to the identification basis in the references (Deng et al., Reference Deng, Wang, Gu and Meng1993), and the mite was identified as Eulaelaps silvestris (Fig. 1). Eulaelaps silvestris’ main distinguishing characteristics are: tritosternum is smaller. The sternal plate anteriorly presents a transversely flattened reticular area with a dense reticulation of inverted teeth. The sternal plate is concave on both the anterior and posterior edges. The genital plate has a deeper notch where it heals with the ventral plate but does not have grooves inward. The anal plate is flat and triangular, with a central bulge on the anterior edge.
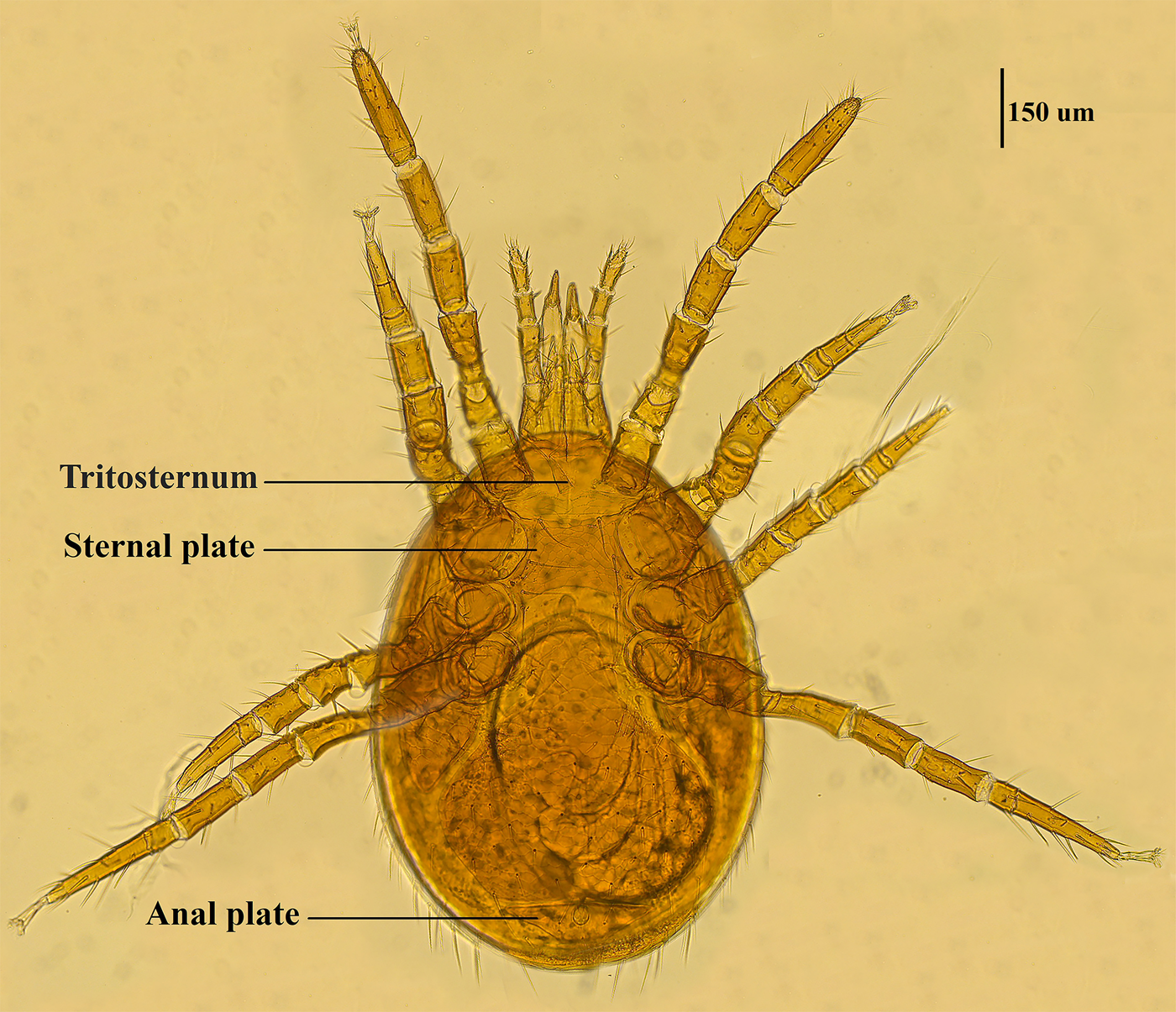
Fig. 1. Morphological characteristics of Eulaelaps silvestris.
DNA extraction and mitochondrial genome sequencing
The adult mites were removed from EP tubes containing 95% ethanol and immersed in sterile distilled water for 30 minutes to remove other microorganisms from the surface, then the mites were cut with a sterile scalpel blade. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, California, USA). The extracted DNA was used to construct Illumina PE libraries. The mitochondrial genome was then sequenced on the Illumina Novoseq 6000 platform (Winnerbio, Shanghai, China). The raw data were processed using Trimmomatic v. 0.35 (Bolger et al., Reference Bolger, Lohse and Usadel2014) to remove low-quality reads and obtain high-quality clean data. Finally, 4.47 GB of clean data were obtained for the assembly of the complete mitochondrial genome of E. silvestris.
Mitochondrial genome annotation
The mitochondrial genome was assembled using MitoZ 2.3 (https://doi.org/10.1101/489955). The MITOS web server predicted genes (Bernt et al., Reference Bernt, Donath, Jühling, Externbrink, Florentz, Fritzsch, Pütz, Middendorf and Stadler2013), and 13 PCGs were compared and edited using the BLAST tool from NCBI. The location and length of tRNA genes were further confirmed using tRNAscan-SE (Lowe and Eddy, Reference Lowe and Eddy1997) and ARWEN (Laslett and Canbäck, Reference Laslett and Canbäck2008). The positions of the 2 rRNA genes were determined based on their putative secondary structures and previously sequenced mitochondrial genomes. The location of the CR was confirmed based on the boundaries of adjacent genes. The mitochondrial sequence of E. silvestris has been deposited in GenBank under the accession number OQ184757. The associated SRA is SRR24658662.
Sequence analysis and phylogenetic reconstruction
Base composition and codon usage were calculated using Geneious v.2020.2 Prime (created by Biomatters; available from https://www.geneious.com) and MEGA X (Kumar et al., Reference Kumar, Tamura and Nei1994), respectively. Calculation of AT-skew and GC-skew was performed according to the formulas [AT-skew = (A – T)/(A + T); GC-skew = (G – C)/(G + C)] (Perna and Kocher, Reference Perna and Kocher1995). The Tandem Repeats Finder Program (http://tandem.bu.edu/trf/trf.html) was used to predict tandem repeat sequences in the CR, and CodonW1.4.2 (https://sourceforge.net/projects/codonw/) was used to determine the relative synonymous codon usage (RSCU) of the 13 PCGs. Limulus polyphemus (JX983598) and Carcinoscorpius rotundicaud (MW446894) were selected as outgroups, and phylogenetic trees were constructed using MrBayes 3.2.7 (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001) and IQ-TREE (Nguyen et al., Reference Nguyen, Schmidt, Von Haeseler and Minh2015) using the Bayesian inference (BI) method and maximum likelihood (ML) method, respectively. Sequence alignment was performed using MAFFT (Katoh et al., Reference Katoh, Misawa, Kuma and Miyata2002). According to the Bayesian information criterion, ModelFinder (Kalyaanamoorthy et al., Reference Kalyaanamoorthy, Minh, Wong, Haeseler and Jermiin2017) was used to determine the best nucleotide substitution model for constructing ML and BI trees (Supplementary materials 1 and 2). Bayesian trees were run for a total of 1 000 000 generations and sampling every 1000 generations; 4 independent Monte Carlo Markov chains were run simultaneously. The first 25% of the trees were burned to ensure sample independence. To estimate the support of the BI tree, the posterior probability (PP) was calculated. For the ML tree, 50 000 ultra-fast bootstrap replications were used to calculate branch reliability (bootstrap probability, BP). The constructed phylogenetic trees were viewed and edited using FigTree 1.4.4 (https://github.com/rambaut/figtree/). The species and accession numbers for the constructed phylogenetic tree are shown in Supplementary material 3.
Results
Mitochondrial genome characteristics of E. silvestris
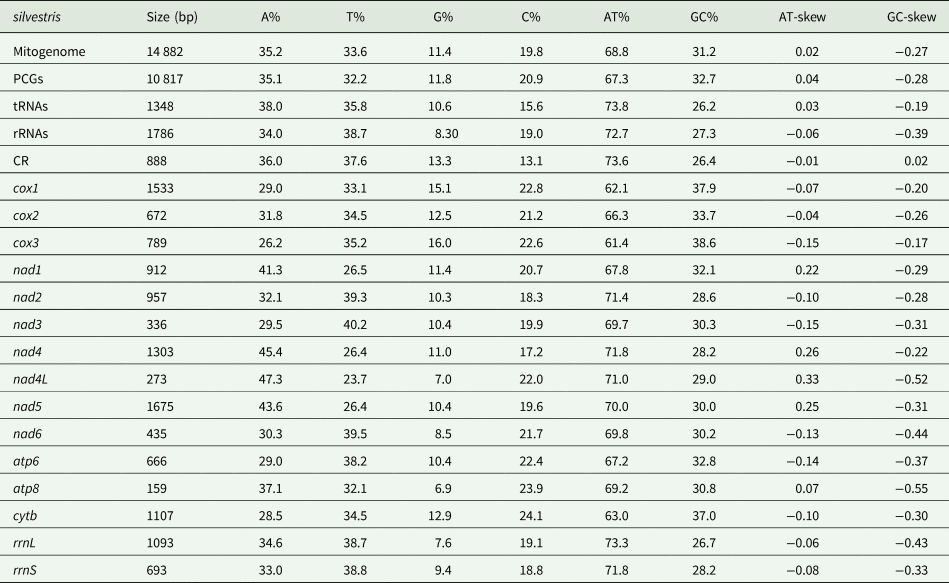
The mitochondrial genome of E. silvestris is a double-stranded DNA molecule with a length of 14 882 bp, consisting of 13 PCG genes (cox1-3, nad1-6, nad4L, atp6, atp8, cytb), 22 tRNA genes, 2 rRNA genes (rrnS and rrnL) and 2 CR (Fig. 2, Table 1). Of the 37 genes, 23 genes (cox1-3, nad2-3, nad6, atp6, atp8, cytb, trnA, trnF, trnD, trnL1, trnG, trnI, trnK, trnM, trnN, trnR, trnS1, trnS2, trnT, trnW) encode on the J-strand. The remaining 14 genes (nad1, nad4, nad4L, nad5, trnC, trnH, trnY, trnE, trnP, trnL2, trnV, trnQ, 16S rRNA and 12S rRNA) are encoded on the N-strand. The complete mitochondrial genome of E. silvestris consisted of 5244 adenine (A), 5007 thymine (T), 2943 cytosine (C) and 1688 guanine (G) in proportions of 35.2, 33.6, 19.8 and 11.4%, respectively (Table 2). The A + T (68.8%) content was much higher than the G + C (31.2%) content, with the lowest content of G. The AT-skew for the complete mitochondrial genome of E. silvestris was 0.02 and the GC-skew was −0.27. The AT content of each region showed a trend of tRNAs > CR > rRNAs > PCGs (Table 2).
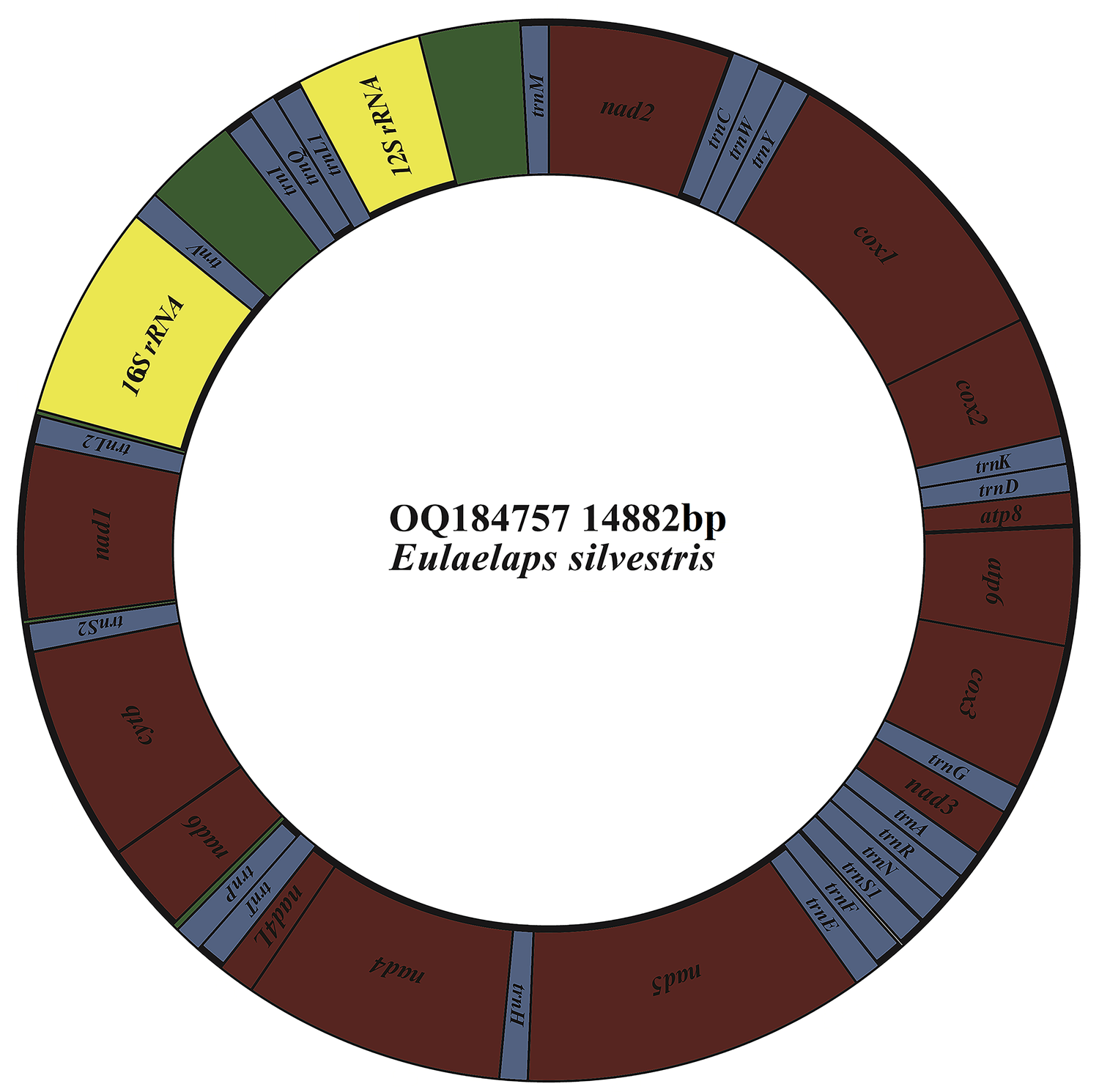
Fig. 2. Mitochondrial genome loop map of Eulaelaps silvestris.
Table 1. Organization of the Eulaelaps silvestris mitochondrial genome
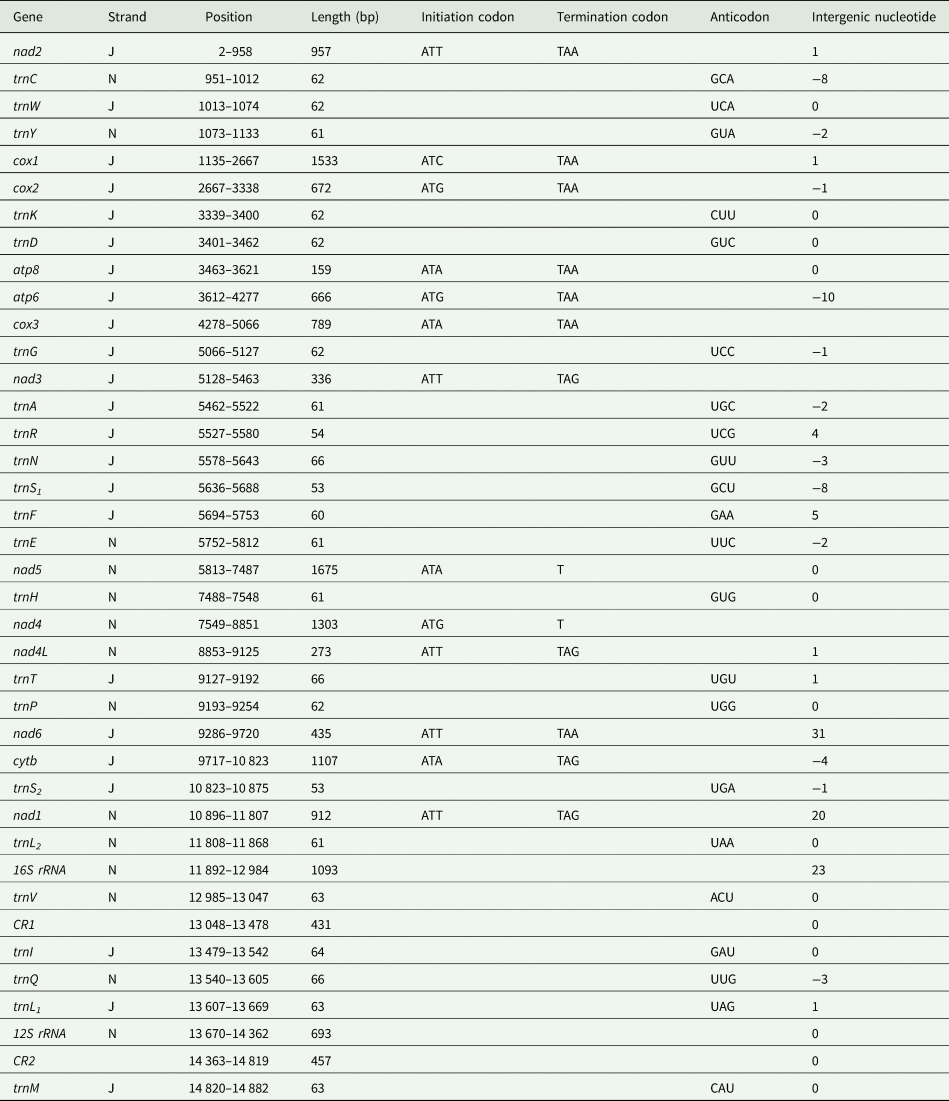
Table 2. Base composition in the Eulaelaps silvestris mitochondrial genome
The complete mitochondrial genome of E. silvestris contains 10 gene inter-genic regions and 12 gene overlap regions (Table 1), with a total length of 91 bp (ranging from 1 to 31 bp) and 45 bp (ranging from 1 to 10 bp), respectively. Intergenic regions were present at 20 gene junctions. The longest intergenic sequence (31 bp) occurs between trnP and nad6; the shortest intergenic sequence (1 bp) occurs between trnY and cox1, nad4 and nad4L, nad4L and trnT, trnQ and trnL1, trnM and nad2. Overlapping regions were present at 24 gene junctions. The longest overlapping sequence (10 bp) occurs between atp8 and atp6; the shortest overlapping sequence (1 bp) occurs between cox1 and cox2, cox3 and trnG, cytb and trnS2 (Table 1). The other 26 gene junctions were without gene intragenic and overlapping regions.
Protein-coding genes and relative synonymous codon usage
The 13 PCGs of E. silvestris had a total length of 10 817 bp, representing 72.7% of the length of the complete mitochondrial genome. AT content was 67.3%, similar to the AT content of the complete mitochondrial genome (Table 1). The shortest and longest genes were atp8 (159 bp) and nad5 (1,675 bp), respectively. All PCGs started with the typical ATN as an initiation codon (ATT: 5, ATA: 4, ATG: 3, ATC: 1), and no rare initiation codons (GTG and TTG) were present. In the use of termination codons, 11 PCGs (cox1-3, nad1, nad2, nad3, nad4L, nad6, atp6, atp8, cytb) ended with the complete termination codons TAA/TAG (TAA: 6, TAG: 4), and only 2 PCGs (nad4 and nad5) had the incomplete termination codons T. The AT-skew of 13 PCGs was at -0.15 (nad3) to 0.33 (nad4L) and the GC-skew at –0.55 (atp8) to –0.17 (cox3). The AT content of individual protein genes was over 60%, and the highest AT content was found in nad2, nad4 and nad4L, while the lowest AT content was found in cox1 and cox3 genes. Meanwhile, there were overlaps between PCGs, such as 1 bp between cox1 and cox2, 4 bp between nad6 and cytb genes, and 10 bp between atp8 and atp6 (Table 1).
The RSCU values for the 13 PCGs of E. silvestris were further analysed and are shown in Fig. 3. The preference for relative synonymous codon usage is related to a number of factors, such as mutational pressure, gene length, gene function, natural selection, etc. (Behura and Severson, Reference Behura and Severson2013; Chen et al., Reference Chen, Shi, Deng, Gu, Xu, Ou, Jiang, Jiao, Zou and Wang2014). RSCU values indicate strong codon usage preference (RSCU > 1), no preference (RSCU = 1) and weak preference (RSCU < 1) (Gupta and Singh, Reference Gupta and Singh2021). In addition to the termination codons, the 13 PCGs contain a total of 3594 codons (Supplementary material 4), which are within the codon number of arthropods (3585–3746) (Cha et al., Reference Cha, Yoon, Lee, Yoon, Hwang, Jin, Han and Kim2007). Phenylalanine (Phe), isoleucine (Ile) and leucine (Leu1) are the most common amino acids, while cysteine (Cys), arginine (Arg), glutamine (Gln) and aspartic acid (Asp) are the less common amino acids. From Fig. 3, the 5 most frequently used codons in the E. silvestris PCGs are UUA (Leu): 2.35, AGA (Ser): 2.08, CCU (Pro): 2.06, UCU (Ser): 2.04, CGA (Arg): 1.76, and all end in A/U. Eulaelaps silvestris mitochondrial PCGs with RSCU > 1 codons preferentially end in A/U; while codons with RSCU < 1 mostly end in G/C, with the exception of GGG (Gly), which has an RSCU of 1.20.
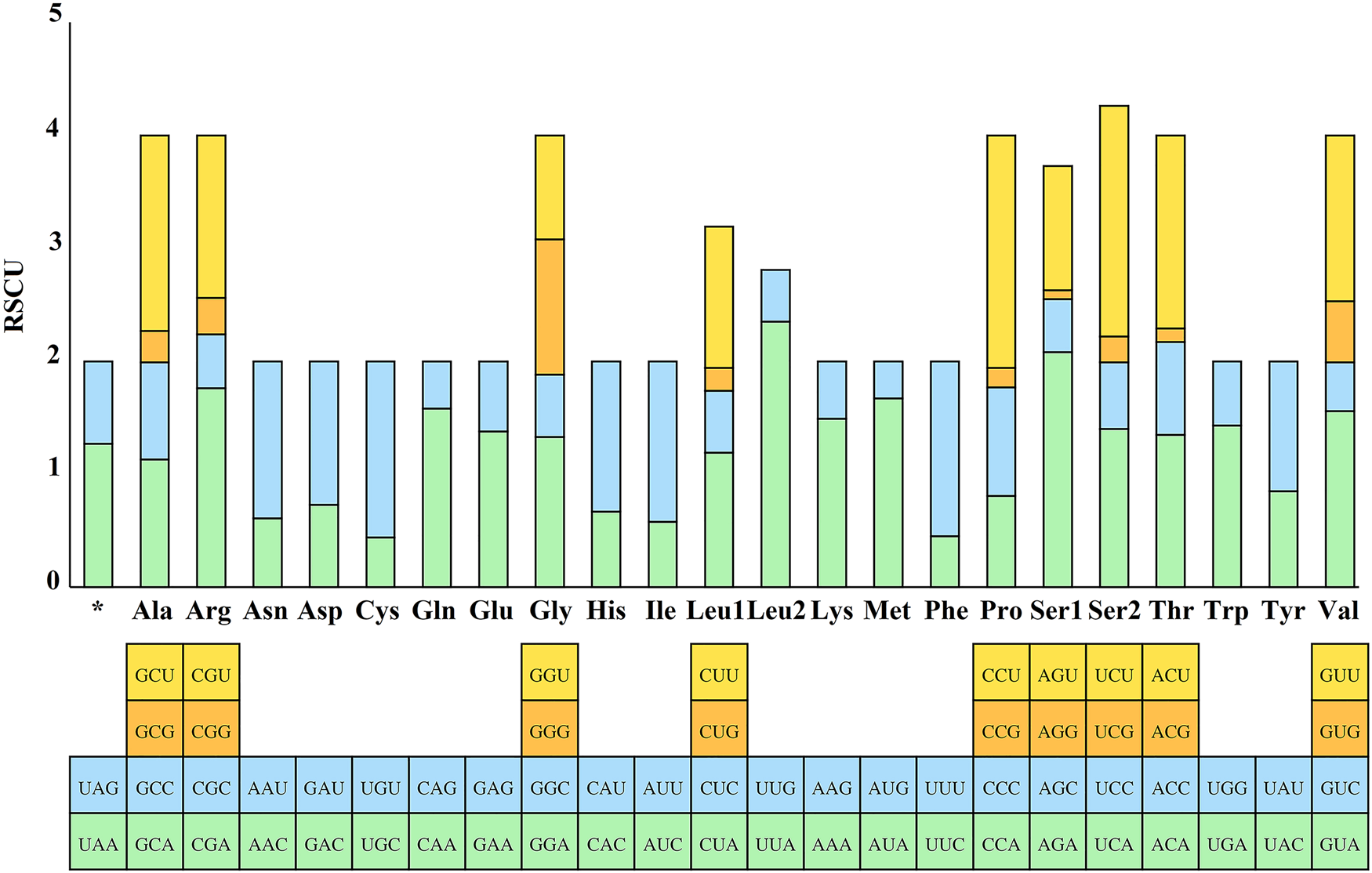
Fig. 3. Relative synonymous codon usage (RSCU) of Eulaelaps silvestris. The Y-axis indicates the RSCU value, and the X-axis indicates the codons corresponding to the respective amino acids.
Transfer RNA, ribosomal RNA genes and control regions
The total length of the 22 tRNA genes in E. silvestris was 1348 bp, with a total AT content of 73.8%, a positive AT-skew (0.03) and a negative GC-skew (−0.19). The length of individual tRNA genes ranged from 53 bp (trnS1 and trnS2) to 66 bp (trnT, trnQ and trnN), and the secondary structures of the 22 tRNA genes are shown in Fig. 4. All of the other 20 tRNA genes are capable of forming a typical cloverleaf secondary structure, except for trnS1 and trnS2, which lack the D arm and cannot form a typical cloverleaf secondary structure. The anticodon arms of the 22 tRNA genes in E. silvestris consist of 4 (trnL1 only) to 5 nucleotide pairs, the amino acid acceptor arms consist of 5 (trnR only) to 7 nucleotide pairs, and the anticodon loops are all composed of 7 nucleotides. In addition to the typical Watson–Crick pairings (A–U, G–C), a total of 38 base mismatches occurred in the E. silvestris mitochondrial genome for 22 tRNA genes during the folding process, including G–U mismatches (19), U–U mismatches (10), U–C mismatches (4), A–A mismatches (3) and A–C mismatches (2). Among them, trnH had the most G–U mismatches (4), trnN, trnV and trnY had the most U–U mismatches (2), trnV had the most U–C mismatches (4), trnV had the most A–A mismatches (3) and trnR and trnV had 1 A–C mismatch each.
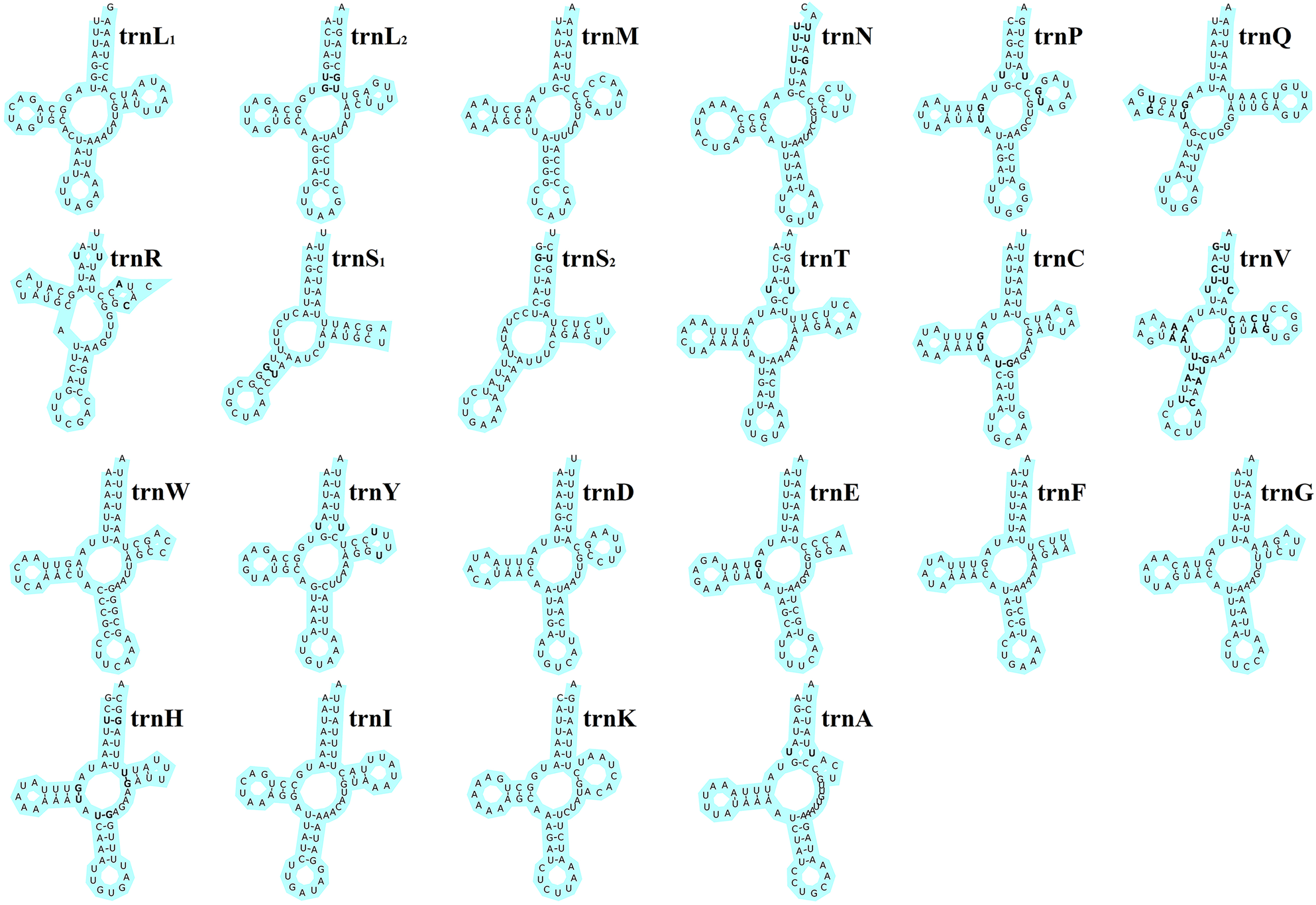
Fig. 4. Eulaelaps silvestris tRNA gene putative secondary structure. Bold indicates mismatch.
Eulaelaps silvestris’ 2 rRNA genes (16S rRNA and 12S rRNA) are 1096 bp and 693 bp in size, respectively (Table 1). The 16S rRNA and 12S rRNA are encoded by the N-strand, same as the 2 rRNA genes of most Acari and arthropods. The 16S rRNA gene was located between trnL2 and trnV with 73.3% AT content, while the 12S rRNA gene was located between trnL1 and CR2 with 71.8% AT content, which was slightly lower than the AT content of the 16S rRNA gene. Duplicated control regions (CR1 and CR2) were identified in the E. silvestris mitochondrial genome. CR1 and CR2 are 431 and 457 bp in length, respectively, and share a common identical core sequence of 308 bp (positions 13 135–13 442 for CR1 and 14 468–14 775 for CR2). CR1 is located between trnV and trnI, and CR2 is located between rrnS and trnM. In the first CR, 2.4 tandem repeats were present, and no tandem repeats were found in the second CR.
Gene rearrangement and phylogenetic analysis
The arrangement order of the mitochondrial genome reflects, to some extent, information on the molecular evolution of mitochondria and is potentially valuable in resolving certain controversial phylogenetic relationships (Tan et al., Reference Tan, Gan, Lee, Linton, Grandjean, Bartholomei-Santos, Miller and Austin2018; Zhang et al., Reference Zhang, Xing, Cheng, Pan, Lv, Cumberlidge and Sun2020). The rearrangement patterns of Mesostigmata sequenced species were analysed using the mitochondrial genome arrangement patterns of hypothetical arthropod ancestors (L. polyphemus) as a reference (Staton et al., Reference Staton, Daehler and Brown1997) (Fig. 5). Among Mesostigmata, only species in the families Parasitidae and Diplogyniidae had the same mitochondrial genome arrangement pattern as that of hypothetical arthropod ancestors, while in the remaining 9 families (Varroidae, Ologamasidae, Dermanyssidae, Laelapidae, Haemogamasidae, Blattisociidae, Rhinonyssidae, Macrochelidae and Phytoseiidae), 20 species of mites were rearranged to different degrees. More importantly, 5 families (Rhinonyssidae, Blattisociidae, Laelapidae, Macrochelidae and Phytoseiidae) were also found to share the same gene clusters among species within families or genera, i.e. Rhinonyssidae intra-family species share 1 gene cluster (trnC-trnS2-trnY-nad1-trnL2-trnL1-trnQ) (note: underlined genes and non-underlined genes indicate that the genes are located on different DNA strands); species of the genera Colaelaps and Hypoaspis within the family Laelapidae share 2 gene clusters (rrnL-trnV-trnM-rrnS and cob-nad2-trnI-trnL1-nad1-trnS2-trnW-trnP-trnY-trnL2-trnQ); species within the genus Blattisocius in the family Blattisociidae share 1 gene (trnI-trnM-nad2-trnW-trnD); species within the genus Macrocheles in the family Macrochelidae share 2 gene clusters (nad4L-trnP-cob and trnT-nad6-trnI-trnQ-trnM); 3 species in different genera of the family Phytoseiidae (Phytoseiulus persimilis, Euseius nicholsi and Amblyseius tsugawai) share 1 gene cluster (cox1-cox2-trnR-nad5-atp6-atp8), and interestingly, P. persimilis and E. nicholsi also share 1 gene cluster (nad4L-nad4-trnD-trnM-trnI-trnK-nad3) and there are 2 additional gene clusters (nad4-trnM-trnI-nad3-cob-trnF-trnQ-trnE-trnG-cox3-trnC and trnL2-nad4L-trnD-trnK-trnN-trnP-trnS2-nad1-trnL1) shared by species of the genus Amblyseius. In this study, E. silvestris is a new rearrangement pattern in Mesostigmata and does not share gene clusters with other families. The rearrangements in E. silvestris occurred mainly in 3 regions, that is, the 3 genes trnL1, rrnS and trnW underwent translocations; the trnF gene underwent inversions; the trnE gene underwent translocations and inversions.

Fig. 5. Mitochondrial genome arrangement pattern of Mesostigmata. Black underline indicates that the gene is located on the N-strand; blue indicates that the gene is inverted and translocated; green indicates that the gene is translocated; orange indicates that the gene is inverted. Two rRNA genes are indicated in dark grey; control region is indicated in light grey. ✱ indicates a duplicated gene; same colour underlines indicate shared gene clusters.
Based on 13 PCGs with L. polyphemus and C. rotundicauda as outgroups, a phylogenetic tree was constructed using the ML method and the BI method (Fig. 6). Both methods obtained a phylogenetic tree with identical topology but different node support. Most branches of the BI tree generally had greater support than the ML tree, and 3 nodes of the ML tree were less than 70. According to the phylogenetic tree results, Mesostigmata is a monophyletic group, and species of the family Diplogyniidae are the early differentiated taxa in Mesostigmata. Most of the mites of the same taxonomic order cluster separately into 1 group, except for the family Laelapidae. In the present study, E. silvestris and species of the family Dermanyssidae clustered together to form a sister branch.
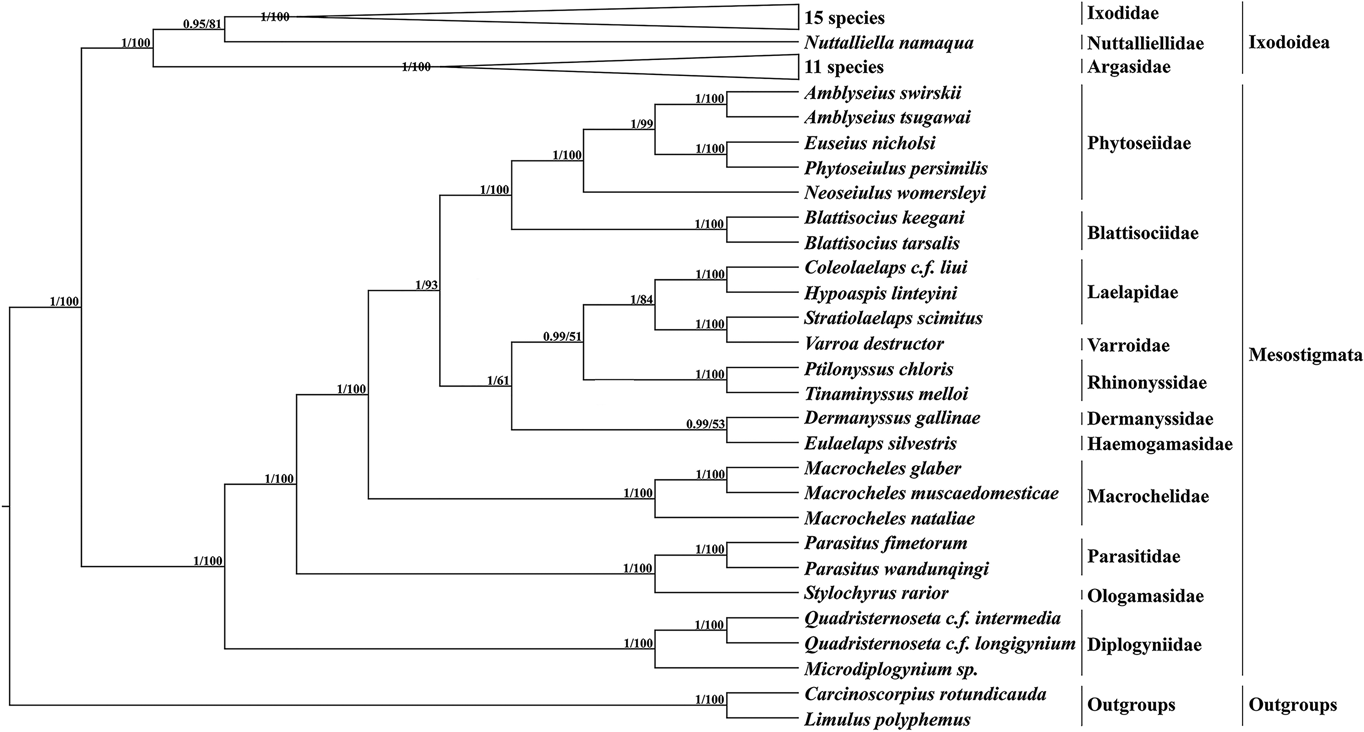
Fig. 6. Phylogenetic tree constructed based on 13 protein-coding genes. The numbers beside the nodes are posterior probabilities (BI) and bootstrap (ML), respectively.
Discussion
In this study, the complete mitochondrial genome of E. silvestris was sequenced and analysed. The mitochondrial genome of E. silvestris, like that of other mites, is a double-stranded DNA molecule containing 37 genes, with a total length of 14 885 bp within the range of the mitochondrial genomes in Mesostigmata sequenced species (Li et al., Reference Li, Shao, Zhang, Deng and Xue2019). It is thought that the main cause of differences in mitochondrial genome size is the number and length of CR, which are positively correlated with species’ mitochondrial genome size, and that differences in the number and length of CR can occur between species (Zhang and Hewitt, Reference Zhang and Hewitt1997). Of the reported mitochondrial genomes of Mesostigmata mites so far, Metaseiulus occidentalis has the largest mitochondrial genome (24 961 bp), but does not possess the greatest number or length of CR; in contrast, Blattisocius tarsalis (16 454 bp) has the greatest number and length of CR, but not the largest mitochondrial genome; Quadristernoseta c. f. intermedia has the smallest mitochondrial genome (14 423 bp), and the CR was not the shortest in length (Li et al., Reference Li, Shao, Zhang, Deng and Xue2019). It can be inferred that the mitochondrial genome sequence length differences in Mesostigmata mites are not significantly correlated with the number and length of CR. Tandem repeats are only present in the first CR of E. silvestris. Tandem repeats have been found only in the CR of the Varroa destructor mitochondrial genome (Navajas et al., Reference Navajas, Le Conte, Solignac, Cros-Arteil and Cornuet2002). It is speculated that tandem repeats may be associated with individual differences in species or life activity levels.
The AT content in the complete mitochondrial genome of E. silvestris was 68.8%, which was lower than the average A + T content of the Acari mitochondrial genome (75.34 ± 4.78%) (Yuan et al., Reference Yuan, Wei, Wang, Dou and Wang2010). The regions with the highest AT content are tRNA genes, and the regions with the lowest AT content are PCGs. It is generally believed that the low AT content of PCGs facilitates the function of encoding the required proteins (Monroe et al., Reference Monroe, Srikant, Carbonell-Bejerano, Becker, Lensink, Exposito-Alonso, Klein, Hildebrandt, Neumann, Kliebenstein, Weng, Imbert, Agren, Rutter, Fenster and Weigel2022). The strand skew of the total nucleotide composition, PCGs and tRAN genes was the same as that of metazoa (positive for AT-skew and negative for GC-skew) (Hassanin et al., Reference Hassanin, Leger and Deutsch2005), while the rRNA genes had negative values for both AT-skew and GC-skew, indicating that the total nucleotide composition, PCGs and tRAN genes were AT-skewed and the rRNA genes were TA-skewed. The nucleotide composition strand bias is associated with asymmetric mutational and selective pressures on the 4 bases during replication and transcription (Hassanin et al., Reference Hassanin, Leger and Deutsch2005). Thus, the nucleotide composition preference in the E. silvestris mitochondrial genome is an important reference for subsequent studies on the replication and transcriptional mechanisms of mitochondrial genomes in species of the family Haemogamasidae.
One of the notable characteristics of the metazoan mitochondrial genome is its more compact intergenic structure. The E. silvestris mitochondrial genome has a total of 10 intergenic regions. The presence of intergenic genes generally promotes organismal evolution because intergenic genes facilitate the storage of more genetic information and increase the probability of mutation. Boyce et al. (Reference Boyce, Zwick and Aquadro1989) suggested that the overlap phenomenon occurring in the mitochondrial genome facilitates the miniaturization of the mitochondrial genome, and that miniaturized mitochondrial genomes have an advantage in natural selection because the time required for their replication is greatly reduced. The E. silvestris mitochondrial genome has a total of 12 gene overlap regions that are shorter than the length of the gene intergenic regions. The gene intergenic regions and gene overlap regions that occur in the mitochondrial genome increase the stability of the mitochondrial structure. Some of the metazoan 13 PCGs exist overlapping each other; for example, the nad4 and nad4L, atp8 and atp6 genes are often found to have an overlap of about 7 bp at their neighboring junctions in the mitochondrial genomes of some insects and ticks (Black and Roehrdanz, Reference Black and Roehrdanz1998; Li et al., Reference Li, Yan and Li2023). In this study, the 13 PCG junctions of E. silvestris only overlap by 10 and 4 bp between atp8 and atp6 and between nad6 and cytb, respectively. The 10 bp overlap between the atp8 and atp6 genes has been widely reported in the mitochondrial genomes of some bird species (Desjardins and Morais, Reference Desjardins and Morais1991; Nishibori et al., Reference Nishibori, Tsudzuki, Hayashi, Yamamoto and Yasue2002).
Compared to the nuclear genome, animal mitochondrial genomes have their own characteristics for the use of codons in the 13 PCGs (Liu and Schultz, Reference Liu and Schultz2010), with most genes starting with ATN and some rare initiation codons, such as TTG (Zhang et al., Reference Zhang, Xu, Wang, Liang, Li and Zhang2022), GTG (Pavan-Kumar et al., Reference Pavan-Kumar, Varshney, Suman, Das, Chaudhari and Krishna2022), CGA (Chen et al., Reference Chen, Zhang, Jin, Yang, Li and Liu2022a), etc. Eulaelaps silvestris’ 13 PCGs were all initiated with a typical ATN, and no rare initiation codons were present. Only 2 PCGs (nad4 and nad5) ended with incomplete termination codons T, whereas the other 11 PCGs had complete termination codons. Incomplete termination codons are common in animal mitochondrial genomes, and it has been demonstrated that these incomplete termination codons can be modified into complete termination codons by polyadenylation of the 3′ end during mRNA processing (Huang et al., Reference Huang, Chen, Wei and Shi2022). Among the 13 PCG codons used, codons ending in A/U were more frequent than those ending in G/C, which may be one of the important reasons for the distinct AT preference in the E. silvestris mitochondrial genome base composition.
Metazoa mitochondrial genome tRNA gene is a cloverleaf secondary structure containing 4 arms and 4 loops, namely amino acid acceptor arm (AA-arm), dihydrouracil arm (DHU) and loop (D-arm and D-loop), anticodon arm and loop (AC-arm and AC-loop), pseudouracil arm and loop (TΨC-arm and TΨC-loop) and a variable loop (Zhang et al., Reference Zhang, Cheng and Ge2019). Almost all animals trnS1 lack the D arm, which is thought to be an ancestral characteristic of metazoa (Xue et al., Reference Xue, Deng, Qu, Hong and Shao2018). In the 22 tRNA genes of E. silvestris, besides trnS1, which lacks the D arm, there is trnS2 which has an atypical cloverleaf secondary structure (missing the D arm). The length of individual tRNA genes of E. silvestris ranged from 53 bp (trnS1 and trnS2) to 66 bp (trnT, trnQ and trnN), with some tRNA genes within the average length (62.0 ± 1.3 bp) of tRNA genes of Parasitiformes species (Yuan et al., Reference Yuan, Wei, Wang, Dou and Wang2010). Eulaelaps silvestris 22 tRNA genes showed a total of 38 times of irregular pairing during the folding process. Among them, G–U mismatches occurred most frequently. It has been demonstrated that the base mismatches occurring in tRNA genes can be corrected by RNA editing without affecting the normal amino acid translocation function (Yokobori and Pääbo, Reference Yokobori and Pääbo1995; Lavrov et al., Reference Lavrov, Brown and Boore2000).
The mitochondrial genome contains 37 genes, which can be arranged in various orders and alignments, and can be used to infer the degree of evolution and relatedness of species by having the same gene order and alignment (Boore and Brown, Reference Boore and Brown1994). In Mesostigmata, species from only 2 families (families Parasitidae and Diplogyniidae) share the same pattern of mitochondrial genome arrangement as the hypothetical arthropod ancestors, while the mitochondrial genomes of the remaining 9 families and 20 species have been rearranged to different degrees, overturning the previous conclusion that the mitochondrial genome arrangement and structure are abnormally stable. Some of the species in the same family or genus have the same gene clusters, suggesting that these identical gene clusters are derivations common to these families or genera. In the E. silvestris mitochondrial genome, only 5 genes have undergone rearrangements, and the rearrangements occurred mainly near tRNA genes and CR. It has been confirmed that the vicinity of tRNA genes and CR are hotspot regions where rearrangements occur, as replication of CR may lead to rearrangements (Kurabayashi et al., Reference Kurabayashi, Sumida, Yonekawa, Glaw, Vences and Hasegawa2008; Yang et al., Reference Yang, Chen and Dong2023). Thus, these rearranged genes or regions may become hotspots for mitochondrial genome research in the future.
The family Haemogamasidae is sometimes considered as a subfamily of the family Laelapidae, but this view has not been shared by most scholars (Vinarski and Korallo-Vinarskaya, Reference Vinarski and Korallo-Vinarskaya2017). Therefore, in this study, based on 13 PCGs, ML and BI trees were constructed, and both phylogenetic trees formed consistent topologies but had slightly different node supports. Both phylogenetic trees support Mesostigmata as a monophyletic group, consistent with the results of Li et al. (Reference Li, Shao, Zhang, Deng and Xue2019), but inconsistent with the conclusion of Lindquist et al. (Reference Lindquist, Krantz, Walter, Krantz and Walter2009b) that Mesostigmata is a polyphyletic group. With the exception of the family Laelapidae, the remaining related species clustered well together according to family order, indicating that the mesostigmata phylogenetic relationships are clear for most species within the family. In this study, E. silvestris formed a sister branch with species from the family Dermanyssidae, indicating that the family Haemogamasidae has the closest affinity to the family Dermanyssidae in Mesostigmata. Meanwhile, the phylogenetic analysis provided strong molecular corroboration that the family Haemogamasidae does not belong to the subfamily Laelapidae.
Conclusion
In this study, the mitochondrial genome of E. silvestris, which is parasitic on the body surface of A. chevrieri, was sequenced and assembled, filling a gap in our understanding of the molecular evolution of species in the genus Eulaelaps. The results showed that E. silvestris is a double-stranded DNA molecule with a size of 14 882 bp. The intergenic arrangement is relatively compact, and the base composition shows a distinct AT preference. The mitochondrial genome of E. silvestris underwent a smaller rearrangement compared to the mitochondrial genome arrangement pattern of arthropod hypothetical ancestors. Phylogenetic analysis showed that the family Haemogamasidae is most closely related to the family Dermanyssidae. The results accumulate molecular data for the genus Eulaelaps and provide a theoretical basis for further studies on the phylogenetic relationships of the genus Eulaelaps.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182023000616.
Data availability
The complete mitochondrial genome sequence of Eulaelaps silvestris is available at the National Center for Biotechnology Information (NCBI) at [https://www.ncbi.nlm.nih.gov/] under accession number OQ184757. The associated SRA is SRR24658662.
Acknowledgements
We thank Chengfu Zhao for his help during specimen collection.
Author contributions
Hui-Juan Yang and Wen-Ge Dong conceived and designed the study. Hui-Juan Yang and Tian-Guang Ren conducted data gathering. Hui-Juan Yang and Zhi-Hua Yang performed statistical analyses. Hui-Juan Yang, Tian-Guang Ren and Zhi-Hua Yang wrote the manuscript. Wen-Ge Dong revised the manuscript.
Financial support
This work was supported by the National Natural Science Foundation of China [No. 32060143 and 31660314 to Wen-Ge Dong].
Competing interest
None.
Ethical standards
Small mammal capture protocols and procedures were approved by the animal ethics committees at Dali University. The approval ID is MECDU-201806-11.











 Open access
Open access