


INTRODUCTION
The trypanosomatids belong to the order Kinetoplastida and include the parasitic protozoa Trypanosoma and Leishmania (the latter are considered elsewhere in this special issue of Parasitology). The trypanosomes represent a significant threat to human and animal health, primarily impacting populations in sub-Saharan Africa and Latin America. There is a limited set of drugs available to treat the diseases caused by these parasites (Wilkinson and Kelly, Reference Wilkinson and Kelly2009; Simarro et al. Reference Simarro, Franco, Diarra, Postigo and Jannin2012b), few new drugs in development and evidence that drug resistance among both animal and human-infective trypanosomes is a significant problem (Geerts et al. Reference Geerts, Holmes, Eisler and Diall2001; Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011). Therefore, understanding the mechanisms by which parasites can and have become resistant to the available drugs is of paramount importance. This review presents the approaches used to dissect the mechanisms of drug resistance in trypanosomes, and how recently developed high throughput techniques are contributing to this process and identifying factors responsible for drug efficacy.
African trypanosomiasis
The African trypanosomes, transmitted by tsetse flies, are extracellular parasites responsible for human African trypanosomiasis (HAT) (Brun et al. Reference Brun, Blum, Chappuis and Burri2010). They have a broad host range, also causing disease in other mammals. Infections with Trypanosoma congolense, T. vivax and T. brucei in wild ungulates (‘hoofed mammal’) result in mild symptoms, while domestic livestock suffer a progressive wasting disease known as Nagana, making stock farming challenging in sub-Saharan Africa (Steverding, Reference Steverding2008). HAT is caused by T. brucei gambiense and T. brucei rhodesiense, with the former causing at least 95% of reported cases. Both forms of HAT progress through two stages. In stage one, the parasites spread through the haemo-lymphatic system from the site of the tsetse bite. In stage two, parasites cross the blood brain barrier and establish an infection in the central nervous system (CNS), typically causing death if the patient remains untreated (Brun et al. Reference Brun, Blum, Chappuis and Burri2010). However, a recent report demonstrated parasite clearance and declining serological response in some individuals even in the absence of treatment, though the prevalence of this apparent trypanotolerance in the human population is unknown (Jamonneau et al. Reference Jamonneau, Ilboudo, Kabore, Kaba, Koffi, Solano, Garcia, Courtin, Laveissiere, Lingue, Buscher and Bucheton2012). The latest estimates indicate that more than 69 million people in sub-Saharan Africa live in known HAT-endemic areas, and of these, more than five million are at high risk of contracting HAT (Simarro et al. Reference Simarro, Cecchi, Franco, Paone, Fevre, Diarra, Postigo, Mattioli and Jannin2012a). Increased case surveillance and improved access to anti-HAT drugs over recent years has led to a significant drop in disease incidence (Brun et al. Reference Brun, Blum, Chappuis and Burri2010). Indeed, since 2009 fewer than 10 000 new cases per annum have been reported, with less than 7000 cases reported in 2011 (Simarro et al. Reference Simarro, Cecchi, Franco, Paone, Fevre, Diarra, Postigo, Mattioli and Jannin2012a), though this is likely to be a substantial underestimate due in part to misdiagnosis and limited access to health-care in some areas (Odiit et al. Reference Odiit, Coleman, Liu, Mcdermott, Fevre, Welburn and Woolhouse2005; Matemba et al. Reference Matemba, Fevre, Kibona, Picozzi, Cleaveland, Shaw and Welburn2010; Mumba et al. Reference Mumba, Bohorquez, Messina, Kande, Taylor, Tshefu, Muwonga, Kashamuka, Emch, Tidwell, Buscher and Meshnick2011).
The necessity for highly co-ordinated efforts to control HAT means disease incidence is vulnerable to the impact of civil conflict and the subsequent breakdown of health-care infrastructure and control programmes; disease incidence reached a historical low in the 1960s, but the cessation of surveillance activities and the outbreak of post-independence conflicts saw a resurgence in HAT (Berrang Ford, Reference Berrang Ford2007), peaking in the late 1990s (Brun et al. Reference Brun, Blum, Chappuis and Burri2010). Recent findings suggest that the current decline in HAT incidence has already led to a concomitant decrease in control efforts in some areas, which could lead to disease resurgence (Ruiz-Postigo et al. Reference Ruiz-Postigo, Franco, Lado and Simarro2012). The downward trend is also vulnerable to the development and spread of parasite resistance to the available drugs.
Chagas disease
Trypanosoma cruzi is the causative agent of Chagas disease, the most important parasitic infection in the Americas. Blood-sucking triatomine bugs are the main vectors of transmission, although ingestion of contaminated food and drink, organ transplantation, blood transfusion and the congenital route also result in a significant number of cases. More than 10 million people in Latin America are estimated to be infected with T. cruzi, resulting in 10–20 000 deaths annually (Moncayo and Silveira, Reference Moncayo and Silveira2009). As a result of migration, the disease is also becoming a global public health issue. For example, in the USA there are thought to be 300 000 individuals with undiagnosed Chagas disease (Bern and Montgomery, Reference Bern and Montgomery2009). In Europe, there have been 4000 confirmed cases in the last 10 years, and more than 80 000 people are estimated to be infected (WHO, 2009).
There are three distinct phases to Chagas disease; acute, indeterminate and chronic. The ‘acute’ stage, which occurs 1–3 weeks after infection, is often asymptomatic. However in children, the outcome can be more serious, with death in 5% of diagnosed cases, mainly due to myocarditis or meningoencephalitis. In most instances, parasitaemia is suppressed following the development of a cellular immune response, although this does not give rise to sterile immunity. Individuals in this ‘indeterminate’ stage remain a source of infection throughout their lives. In about 30% of cases, the disease progresses from the ‘indeterminate’ to the ‘chronic’ stage, sometimes decades after the primary infection (Kirchhoff, Reference Kirchhoff2011), leading to clinical outcomes such as cardiomyopathy, alimentary tract pathology (typically, megacolon and megaoesophagus) and/or damage to the peripheral nervous system. In several regions of South America, Chagasic heart disease is a common cause of sudden cardiac failure. Co-infections with HIV can lead to activation of chronic Chagas disease, often with atypical clinical manifestations, including CNS involvement (Diazgranados et al. Reference Diazgranados, Saavedra-Trujillo, Mantilla, Valderrama, Alquichire and Franco-Paredes2009). Chagas disease presents significant challenges in terms of diagnosis, vector control and treatment, and the situation is further complicated by the development of resistance to the available drugs.
CHEMOTHERAPY
It is unlikely that a vaccine will be developed against any human trypanosome in the near future. In the case of HAT, this is primarily due to antigenic variation of the surface coat, enabling T. brucei to evade the immune response until the host succumbs (Horn and McCulloch, Reference Horn and Mcculloch2010). T. cruzi vaccine development shows some promise (Cazorla et al. Reference Cazorla, Frank and Malchiodi2009), but it has been notoriously challenging to develop any anti-protozoal vaccine. Therefore, public health measures have focused on the insect vectors, making a significant contribution to the control of Chagas disease and HAT (Hashimoto and Schofield, Reference Hashimoto and Schofield2012; Welburn and Maudlin, Reference Welburn and Maudlin2012). Despite this, there have been concerns about the sustainability of the programmes. Hence, though limited by issues of specificity, toxicity and developing parasite resistance, chemotherapy is fundamental to the control of African trypanosomiasis and Chagas disease.
African trypanosomiasis
There are five drugs currently in use for the treatment of HAT; their application is dependent upon disease stage and the identity of the infecting subspecies (Table 1). Stage one T. b. gambiense and T. b. rhodesiense HAT are treated with pentamidine or suramin, respectively; both drugs have been in constant use for decades – suramin was developed in 1916 and pentamidine in 1937 (Steverding, Reference Steverding2008). Since 1990 and until recently, eflornithine has been used as a monotherapy to treat stage two T. b. gambiense HAT. It is now recommended to be used as part of NECT (nifurtimox-eflornithine combination therapy), which has equivalent therapeutic outcomes to eflornithine monotherapy, but allows for reduced dosing resulting in greater patient compliance (Priotto et al. Reference Priotto, Kasparian, Mutombo, Ngouama, Ghorashian, Arnold, Ghabri, Baudin, Buard, Kazadi-Kyanza, Ilunga, Mutangala, Pohlig, Schmid, Karunakara, Torreele and Kande2009).
Table 1. Current drugs – their application, and resistance mechanisms identified by low throughput analysis of laboratory-derived and clinical resistant isolates

Notes
a Not determined.
b Following selection, a suramin-resistant phenotype was expressed in bloodstream but not insect stage parasites (Scott et al. Reference Scott, Tait and Turner1996).
c Treatment failures reported in west Africa in the 1950s (Pepin and Milord, Reference Pepin and Milord1994).
d Pentamidine resistant T. brucei can be generated in the laboratory, but they have not been reported in the field (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011).
e Mutation or loss of AT1 renders T. brucei less sensitive to melarsoprol, however not all resistant clinical isolates have modified this locus (Matovu et al. Reference Matovu, Geiser, Schneider, Maser, Enyaru, Kaminsky, Gallati and Seebeck2001).
f Eflornithine monotherapy treatment failures reported in some foci (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011), as well as a limited number of NECT relapses (Franco et al. Reference Franco, Simarro, Diarra, Ruiz-Postigo, Samo and Jannin2012).
Melarsoprol, which has been in use since 1949, is the only drug effective against both forms of HAT during stage two disease, though its use can lead to a devastating reactive encephalitis in 5–10% of cases. This is thought to be due to massive release of parasite antigens and a subsequent autoimmune reaction (Pepin and Milord, Reference Pepin and Milord1994). Until the introduction of eflornithine and NECT for the treatment of T. b. gambiense HAT, melarsoprol was the only drug effective against either late stage HAT. Since these alternative treatments have become available, melarsoprol use has declined, being used in only 12% of reported cases in 2010 (Simarro et al. Reference Simarro, Franco, Diarra, Postigo and Jannin2012b). However, melarsoprol is still the only available treatment for late stage T. b. rhodesiense HAT. All the licensed anti-HAT drugs require administration in hospital and come with significant side effects (Barrett and Croft, Reference Barrett and Croft2012). There may soon be new chemotherapeutic options, with fexnidazole, an orally active nitroimidazole, and the benzoxaborole, SCYX-7158, about to enter phase II and phase I clinical trials, respectively (Jacobs et al. Reference Jacobs, Nare, Wring, Orr, Chen, Sligar, Jenks, Noe, Bowling, Mercer, Rewerts, Gaukel, Owens, Parham, Randolph, Beaudet, Bacchi, Yarlett, Plattner, Freund, Ding, Akama, Zhang, Brun, Kaiser, Scandale and Don2011; Barrett and Croft, Reference Barrett and Croft2012). In addition, novel diamidines are showing early promise in animal models of HAT (Thuita et al. Reference Thuita, Wang, Kagira, Denton, Paine, Mdachi, Murilla, Ching, Boykin, Tidwell, Hall and Brun2012).
As with HAT, the range of drugs for the treatment of Nagana is limited and resistant parasites are widespread. There are three drugs currently used to treat Nagana, all of which have been in use for more than 50 years: homidium bromide and the related isometamidium chloride, and diminazene aceturate (berenil). The widespread and often unregulated use of these drugs, both as treatments and in prophylaxis, has inevitably led to the development of drug resistance (Geerts et al. Reference Geerts, Holmes, Eisler and Diall2001). This is a particularly acute problem in highly endemic areas with very high drug use (Delespaux and de Koning, Reference Delespaux and De Koning2007).
Chagas disease
For more than 40 years, the nitroheterocyclic agents, nifurtimox and benznidazole, have been the front line drugs for the treatment of Chagas disease (Wilkinson and Kelly, Reference Wilkinson and Kelly2009). However, these drugs are far from optimal. They are effective against infections in the acute phase, but their usefulness in preventing or alleviating symptoms in the chronic stage remains controversial (Marin-Neto et al. Reference Marin-Neto, Rassi, Morillo, Avezum, Connolly, Sosa-Estani, Rosas and Yusuf2008; Urbina, Reference Urbina2010). Both drugs have been reported to be carcinogenic and display a wide range of side effects which include CNS toxicity, leukopenia, muscle weakness and severe dermatitis (Castro et al. Reference Castro, De Mecca and Bartel2006; Wilkinson and Kelly, Reference Wilkinson and Kelly2009). This, coupled with treatment regimes that extend over several months, frequently results in a failure to complete the therapeutic schedule. T. cruzi strains refractory to treatment are encountered throughout South America (Castro et al. Reference Castro, De Mecca and Bartel2006), though the extent to which this reflects acquired resistance or natural variation in sensitivity is unknown.
The large number of infected individuals throughout Latin America and beyond makes the development of new therapies to treat Chagas disease a research priority. A major question is whether drugs targeted at the parasite are the best strategy for treating the chronic stage, given the numerous reports that autoimmune responses could be a significant determinant of disease pathology. However, recent studies using murine models have shown that the continued presence of the parasite is both necessary and essential for development of cardiac disease (Tarleton et al. Reference Tarleton, Zhang and Downs1997; Tarleton, Reference Tarleton2003; Garcia et al. Reference Garcia, Ramos, Senra, Vilas-Boas, Rodrigues, Campos-De-Carvalho, Ribeiro-Dos-Santos and Soares2005). Consistent with this, antiparasitic drugs can block chronic cardiomyopathy, and give rise to a stable protective T cell memory (Bustamante et al. Reference Bustamante, Bixby and Tarleton2008). Taken together, these reports suggest that chemotherapeutic intervention is appropriate, even against the chronic stage of the disease.
The research community is taking a twin-track approach to improving therapy against Chagas disease. Firstly, as outlined below, there has been a drive to identify resistance mechanisms so that treatment regimes based on current drugs can be optimized. Secondly, the development of new drugs is being widely pursued (Wilkinson and Kelly, Reference Wilkinson and Kelly2009; Le Loup et al. Reference Le Loup, Pialoux and Lescure2011). Promising drug targets currently under investigation include enzymes of the ergosterol biosynthetic pathway (Buckner et al. Reference Buckner, Yokoyama, Lockman, Aikenhead, Ohkanda, Sadilek, Sebti, Van Voorhis, Hamilton and Gelb2003; Suryadevara et al. Reference Suryadevara, Olepu, Lockman, Ohkanda, Karimi, Verlinde, Kraus, Schoepe, Van Voorhis, Hamilton, Buckner and Gelb2009; Urbina, Reference Urbina2009) and the cathepsin L-like cysteine protease, cruzipain, with several inhibitors at various stages of development (McKerrow et al. Reference McKerrow, Doyle, Engel, Podust, Robertson, Ferreira, Saxton, Arkin, Kerr, Brinen and Craik2009; Beaulieu et al. Reference Beaulieu, Isabel, Fortier, Masse, Mellon, Methot, Ndao, Nicoll-Griffith, Lee, Park and Black2010). Two anti-fungal triazole compounds, posoconazole and E1224 (inhibitors of lanosterol 14 α-demethylase, CYP51), are presently undergoing clinical trials (Barrett and Croft, Reference Barrett and Croft2012).
Are treatment failures associated with drug resistance?
Treatment failures have been reported for all of the currently available HAT monotherapies, though in the case of pentamidine these are rare (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011). Difficulties encountered in the staging of HAT infections and the subsequent administration of chemotherapy, mean that it is often difficult to ascribe these failures to the presence of bona fide drug-resistant parasites. However, data derived from experiments on drug-resistant laboratory strains of T. brucei enabled genotypic analysis of parasites from relapse patients in Uganda, indicating that at least some melarsoprol treatment failures are due to the development of resistance (Matovu et al. Reference Matovu, Geiser, Schneider, Maser, Enyaru, Kaminsky, Gallati and Seebeck2001). Until the 1950s suramin was used to treat T. b. gambiense HAT, since when its use has been restricted to treating T. b. rhodesiense HAT, in part due to a large number of reported treatment failures in West Africa. More recently, there have been a number of anecdotal reports of eflornithine monotherapy treatment failures, though it is not known if these were due to the presence of eflornithine-resistant parasites (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011). NECT has only been in use since 2009, however its use against T. b. gambiense HAT is now widespread, with 59% of all second stage cases treated with this combination therapy in 2010 (Simarro et al. Reference Simarro, Franco, Diarra, Postigo and Jannin2012b), and there are already reports of small numbers of relapses (Franco et al. Reference Franco, Simarro, Diarra, Ruiz-Postigo, Samo and Jannin2012). It is not known whether these were due to the presence of drug-resistant trypanosomes, although this is a possibility, given the ease with which parasites resistant to either drug can be selected in the laboratory (see below).
Understanding how trypanosomes can become resistant to the available drugs enables the development of diagnostic tools for the identification of genuinely resistant parasites in the field. Indeed, genetic assays designed to identify the presence of known determinants of drug resistance have revealed high prevalence of resistance to berenil amongst T. congolense isolated from cattle and game animals in endemic areas of Ethiopia and Southern Africa (Chitanga et al. Reference Chitanga, Marcotty, Namangala, Van Den Bossche, Van Den Abbeele and Delespaux2011; Moti et al. Reference Moti, Fikru, Van Den Abbeele, Buscher, Van Den Bossche, Duchateau and Delespaux2012).
Distinguishing between treatment failure due to drug resistance or for other reasons is even more challenging in the case of Chagas disease. The toxicity of the available drugs in combination with the need for prolonged treatment regimes has an inevitable impact on patient compliance, providing a selective environment for the development of drug resistant parasites (see above).
IDENTIFYING RESISTANCE MECHANISMS – LOW THROUGHPUT APPROACHES
Trypanosomes resistant to each of the licensed HAT and Chagas disease monotherapies have been derived by in vitro or in vivo selection in the laboratory. With the exception of suramin resistance (Scott et al. Reference Scott, Tait and Turner1996), it has been possible to define the underlying mechanism in each case (Table 1). Biochemical assays, inspired by insights into the chemistry of the compound under investigation, have been used to define the character of the resistance mechanism. Using various genetic approaches it has been possible to identify the gene products responsible for some of the observed resistance phenotypes. This broad approach was used to identify the genes encoding the trypanosome P2 adenosine transporter, AT1 (Carter and Fairlamb, Reference Carter, Berger and Fairlamb1993; Maser et al. Reference Maser, Sutterlin, Kralli and Kaminsky1999), and the amino acid transporter, AAT6 (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010), as key determinants of melarsoprol and eflornithine uptake, respectively, in T. brucei. Candidate-based reverse genetic approaches have also identified potential drug resistance determinants, including the melarsoprol-trypanothione (Mel-T) transporter, multidrug resistance protein A (MRPA) (Shahi et al. Reference Shahi, Krauth-Siegel and Clayton2002), and two nucleobase transporters related to AT1, NT11·1 and NT12·1, capable of taking up pentamidine (Ortiz et al. Reference Ortiz, Sanchez, Quecke and Landfear2009). A similar methodology identified the trypanosome nitroreductase (NTR), as the activator of the pro-drugs nifurtimox and benznidazole in T. cruzi (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008). These studies were facilitated by the sequencing of the trypanosome genomes (Berriman et al. Reference Berriman, Ghedin, Hertz-Fowler, Blandin, Renauld, Bartholomeu, Lennard, Caler, Hamlin, Haas, Bohme, Hannick, Aslett, Shallom, Marcello, Hou, Wickstead, Alsmark, Arrowsmith, Atkin, Barron, Bringaud, Brooks, Carrington, Cherevach, Chillingworth, Churcher, Clark, Corton, Cronin, Davies, Doggett, Djikeng, Feldblyum, Field, Fraser, Goodhead, Hance, Harper, Harris, Hauser, Hostetler, Ivens, Jagels, Johnson, Johnson, Jones, Kerhornou, Koo, Larke, Landfear, Larkin, Leech, Line, Lord, Macleod, Mooney, Moule, Martin, Morgan, Mungall, Norbertczak, Ormond, Pai, Peacock, Peterson, Quail, Rabbinowitsch, Rajandream, Reitter, Salzberg, Sanders, Schobel, Sharp, Simmonds, Simpson, Tallon, Turner, Tait, Tivey, Van Aken, Walker, Wanless, Wang, White, White, Whitehead, Woodward, Wortman, Adams, Embley, Gull, Ullu, Barry, Fairlamb, Opperdoes, Barrell, Donelson, Hall, Fraser, Melville and El-Sayed2005; El-Sayed et al. Reference El-Sayed, Myler, Bartholomeu, Nilsson, Aggarwal, Tran, Ghedin, Worthey, Delcher, Blandin, Westenberger, Caler, Cerqueira, Branche, Haas, Anupama, Arner, Aslund, Attipoe, Bontempi, Bringaud, Burton, Cadag, Campbell, Carrington, Crabtree, Darban, Da Silveira, De Jong, Edwards, Englund, Fazelina, Feldblyum, Ferella, Frasch, Gull, Horn, Hou, Huang, Kindlund, Klingbeil, Kluge, Koo, Lacerda, Levin, Lorenzi, Louie, Machado, Mcculloch, Mckenna, Mizuno, Mottram, Nelson, Ochaya, Osoegawa, Pai, Parsons, Pentony, Pettersson, Pop, Ramirez, Rinta, Robertson, Salzberg, Sanchez, Seyler, Sharma, Shetty, Simpson, Sisk, Tammi, Tarleton, Teixeira, Van Aken, Vogt, Ward, Wickstead, Wortman, White, Fraser, Stuart and Andersson2005), the development of reverse genetic tools, such as stable transfection, inducible protein expression and RNA interference (RNAi) systems (Lee and Van der Ploeg, Reference Lee and Van Der Ploeg1990; Wirtz and Clayton, Reference Wirtz and Clayton1995; Wirtz et al. Reference Wirtz, Leal, Ochatt and Cross1999; LaCount et al. Reference Lacount, Bruse, Hill and Donelson2000; Wang et al. Reference Wang, Morris, Drew and Englund2000; Alsford et al. Reference Alsford and Horn2005b; Taylor and Kelly, Reference Taylor and Kelly2006; Alsford and Horn, Reference Alsford, Eckert, Baker, Glover, Sanchez-Flores, Leung, Turner, Field, Berriman and Horn2008), and early forward genetic tools, such as trypanosome cDNA and genomic DNA libraries (Maser et al. Reference Maser, Sutterlin, Kralli and Kaminsky1999; Shahi et al. Reference Shahi, Krauth-Siegel and Clayton2002).
Characterizing melarsoprol uptake and efflux
Melarsoprol is a melaminophenyl arsenical developed in the 1940s (Friedheim, Reference Friedheim1949) and thought to act via the formation of Mel-T, a toxic adduct with the trypanosome-specific thiol, trypanothione (Fairlamb et al. Reference Fairlamb, Henderson and Cerami1989; Alsford et al. Reference Alsford, Turner, Obado, Sanchez-Flores, Glover, Berriman, Hertz-Fowler and Horn2012). Melarsoprol contains a melamine ring, a nitrogen-rich heterocycle with similarities to several natural metabolites, an observation that led to the ‘melamine receptor’ hypothesis (Barrett and Fairlamb, Reference Barrett and Fairlamb1999). Assessing the ability of more than 100 nitrogen-containing biochemicals to inhibit cell lysis by melarsoprol revealed adenine and adenosine as potent inhibitors, and subsequent biochemical characterization identified the P2 adenosine transporter as a mediator of melarsoprol uptake (Carter and Fairlamb, Reference Carter, Berger and Fairlamb1993). This work was done prior to the sequencing of the T. brucei genome, at a time when the identification of the proteins responsible for a particular phenotype could be a laborious process. Mäser and colleagues came up with a creative solution. Saccharomyces cerevisiae is normally incapable of taking up adenosine, so they reasoned that expression of the trypanosome transporter in yeast defective for adenosine biogenesis would enable their survival. The mutant yeast was transformed with a T. brucei cDNA library and grown on media supplemented with adenosine, and cells expressing the trypanosome P2 purine transporter, AT1, were able to grow (Maser et al. Reference Maser, Sutterlin, Kralli and Kaminsky1999).
AT1 was subsequently found to be mutated or absent from a number of melarsoprol-resistant strains (Maser et al. Reference Maser, Sutterlin, Kralli and Kaminsky1999; Matovu et al. Reference Matovu, Geiser, Schneider, Maser, Enyaru, Kaminsky, Gallati and Seebeck2001; Stewart et al. Reference Stewart, Burchmore, Clucas, Hertz-Fowler, Brooks, Tait, MacLeod, Turner, De Koning, Wong and Barrett2010). However, loss of AT1 function was only seen in some isolates from melarsoprol-treated relapse patients (Matovu et al. Reference Matovu, Geiser, Schneider, Maser, Enyaru, Kaminsky, Gallati and Seebeck2001). It was subsequently found that AT1 gene deletion confers only a two-fold decrease in melarsoprol sensitivity (Matovu et al. Reference Matovu, Stewart, Geiser, Brun, Maser, Wallace, Burchmore, Enyaru, Barrett, Kaminsky, Seebeck and De Koning2003), suggesting that an additional or alternative factor must be driving high level resistance. Contemporary work on drug resistance in Leishmania tarentolae identified the ABC transporter, LtMRPA, as contributing to resistance to trivalent antimony via efflux of the metal-trypanothione conjugate (Legare et al. Reference Legare, Richard, Mukhopadhyay, Stierhof, Rosen, Haimeur, Papadopoulou and Ouellette2001). It was proposed that T. brucei might be able to remove Mel-T using a similar transporter. The T. brucei orthologue of LtMRPA was isolated by screening a genomic DNA library with a known T. brucei ABC transporter sequence, and its over-expression led to a ten-fold increase in melarsoprol EC50in vitro (Shahi et al. Reference Shahi, Krauth-Siegel and Clayton2002). However, MRPA over-expression has not been found in melarsoprol-resistant clinical isolates (Alibu et al. Reference Alibu, Richter, Voncken, Marti, Shahi, Renggli, Seebeck, Brun and Clayton2006), so its contribution to treatment failures in the field remains equivocal.
Soon after the identification of the role of AT1 in the uptake of melarsoprol, it was shown that this transporter also contributed to the uptake of pentamidine and other diamidines, such as the anti-nagana drug, berenil (Barrett et al. Reference Barrett, Vincent, Burchmore, Kazibwe and Matovu1995; Carter et al. Reference Carter and Fairlamb1995; de Koning et al. Reference De Koning, Anderson, Stewart, Burchmore, Wallace and Barrett2004). Two other uptake mechanisms distinct from AT1 have been biochemically characterized, the high and low affinity pentamidine transporters (or HAPT and LAPT) (de Koning Reference De Koning2001, Reference De Koning2008; Bridges et al. Reference Bridges, Gould, Nerima, Maser, Burchmore and De Koning2007). The two AT1-related nucleobase transporters, NT11·1 and NT12·1, have been shown to be capable of pentamidine uptake in T. brucei, though neither seems to correspond to HAPT or LAPT (Ortiz et al. Reference Ortiz, Sanchez, Quecke and Landfear2009). Therefore, pentamidine may be able to access the T. brucei interior via multiple routes, possibly explaining why treatment failures with this drug are rare. Those that do occur may arise from factors other than changes in pentamidine uptake by the infecting parasite.
Identifying the eflornithine transporter
Eflornithine, an analogue of ornithine, blocks spermidine synthesis and the formation of trypanothione through the inhibition of ornithine decarboxylase (ODC) (Bacchi et al. Reference Bacchi, Garofalo, Mockenhaupt, Mccann, Diekema, Pegg, Nathan, Mullaney, Chunosoff, Sjoerdsma and Hutner1983), which normally catalyses the conversion of ornithine to putrescine. It is effective against T. b. gambiense, but T. b. rhodesiense is naturally tolerant to eflornithine as a result of several factors; these include reduced drug uptake, increased putrescine uptake or higher ODC turnover, depending on the isolate analysed (Bacchi et al. Reference Bacchi, Garofalo, Ciminelli, Rattendi, Goldberg, McCann and Yarlett1993; Iten et al. Reference Iten, Mett, Evans, Enyaru, Brun and Kaminsky1997). The existence of T. b. gambiense resistant to eflornithine has yet to be confirmed in the field, in spite of recent anecdotal reports of monotherapy treatment failures (Barrett et al. Reference Barrett, Zhang, Denise, Giroud and Baltz2011). It should be noted that eflornithine use has only become widespread since the introduction of NECT (Priotto et al. Reference Priotto, Kasparian, Mutombo, Ngouama, Ghorashian, Arnold, Ghabri, Baudin, Buard, Kazadi-Kyanza, Ilunga, Mutangala, Pohlig, Schmid, Karunakara, Torreele and Kande2009; Simarro et al. Reference Simarro, Franco, Diarra, Postigo and Jannin2012b). Indeed, eflornithine-resistant trypanosomes can be generated easily in the laboratory, and cells exhibiting significantly reduced drug uptake were derived before the drug was widely used (Phillips and Wang, Reference Phillips and Wang1987). However, it was not until 2010 that the resistance mechanism was defined (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010). By this time, significant advances had been made in the field of metabolomics, enabling the quantification of multiple metabolites through time in treated and untreated cells (Creek et al. Reference Creek, Anderson, McConville and Barrett2012). This led to the observation that, while the levels of spermidine and the intermediates of its synthesis were no different in resistant and sensitive cell lines, eflornithine accumulation was markedly reduced in the resistant line (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010). A systematic analysis of the amino acid transporters in the eflornithine-resistant line revealed that the AAT6 transporter had been lost from these cells. Its key role in eflornithine uptake was confirmed using RNAi knockdown (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010).
The nifurtimox-benznidazole activator
The genetic tools applicable to T. cruzi include a wide range of episomal expression vectors and robust methodology that allows targeted gene deletion (Taylor et al. Reference Taylor, Huang and Kelly2011). However, T. cruzi reverse genetics procedures are generally less flexible than those of T. brucei, mainly due to the length of time required for the isolation of transformed parasites (∼4 weeks as opposed to 5 days) and the lack of RNAi-based systems. T. cruzi is unable to generate an RNAi response since genes encoding the Argonaute and DICER-like proteins are absent (Lye et al. Reference Lye, Owens, Shi, Murta, Vieira, Turco, Tschudi, Ullu and Beverley2010). Evidence suggests they were lost after divergence of the two trypanosome species. Despite these limitations, the available tools have been experimentally crucial in increasing our understanding of T. cruzi infection biology and the mechanisms of drug activity and resistance.
Nifurtimox and benznidazole are nitroheterocyclic compounds in which a nitro-group is linked to a furan and an imidazole, respectively (Fig. 1). Both are pro-drugs, but the specific NTRs required for their activation and the nature of the resulting toxic metabolites had remained unresolved, despite being a major focus of research for more than 40 years. With nifurtimox, early experiments had suggested that trypanocidal activity could arise from the generation of toxic oxygen metabolites, mediated by one-electron reduction of the drug by type II NTR activity (Docampo, Reference Docampo1990; Viode et al. Reference Viode, Bettache, Cenas, Krauth-Siegel, Chauviere, Bakalara and Perie1999). In an aerobic environment, this results in a ‘futile cycling’ process in which NADPH is consumed, superoxide radicals produced and nifurtimox regenerated. However, parasites engineered to have an enhanced oxidative defence capacity were no more resistant to nifurtimox than wild type T. cruzi, suggesting that this process has limited in vivo significance (Taylor et al. Reference Taylor, Huang and Kelly2011).
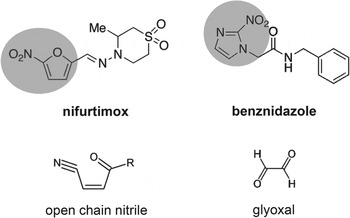
Fig. 1. Structures of the nitroheterocyclic drugs used to treat T. cruzi infections and their toxic metabolites. The highlighted regions of nifurtimox and benznidazole correspond to the 5-nitrofuran and the 2-nitroimidazole groups, respectively. The structures of the toxic unsaturated open chain nitrile and glyoxal metabolites (Hall et al. Reference Hall and Wilkinson2011; Hall and Wilkinson, Reference Hall, Bot and Wilkinson2012) are shown below the corresponding drugs.
In bacteria, resistance to nitrofuran pro-drugs results from mutations in the flavin-dependent oxidoreductases of the type I NTR family which catalyse the O2-insensitive NAD(P)H-dependent two-electron reduction of the nitro group (Whiteway et al. Reference Whiteway, Koziarz, Veall, Sandhu, Kumar, Hoecher and Lambert1998; Parkinson et al. Reference Parkinson, Skelly and Neidle2000). This results in a hydroxylamine product, and ultimately the generation of nitrenium ions, which can cause lesions in chromosomal DNA and damage to other biological molecules (McCalla et al. Reference McCalla, Reuvers and Kaiser1971; Streeter and Hoener, Reference Streeter and Hoener1988). A type I NTR-like protein was identified in the T. cruzi genome (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008). Biochemical analysis demonstrated that TcNTR could reduce nifurtimox, benznidazole and other nitroheterocyclic drugs in an NADH-dependent manner. TcNTR-mediated nifurtimox reduction leads to the production of an unsaturated open-chain nitrile (Fig. 1), the metabolite responsible for the trypanocidal activity (Hall et al. Reference Hall and Wilkinson2011). With benznidazole, reductive metabolism leads to the formation of glyoxal (Fig. 1), which has a range of cytotoxic properties (Hall and Wilkinson, Reference Hall, Bot and Wilkinson2012). Deletion of one copy of TcNTR results in 2–5 fold resistance against a range of nitroheterocyclics, presumably because of the reduced rate of drug metabolism (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008). The observation that heterozygotes do not display reduced infectivity, suggests a possible route for the development of acquired cross-resistance to this class of drugs. Further experimentation demonstrated that TcNTR null mutants have a much reduced capacity to infect mammalian cells and to divide as amastigotes, indicating an upper limit to clinically relevant resistance by this mechanism (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008; Mejia et al. Reference Mejia, Hall, Taylor, Gomez-Palacio, Wilkinson, Triana-Chavez and Kelly2012).
T. cruzi cross-resistant to nitroheterocyclic drugs are readily selectable in the laboratory. This can result from the loss of one copy of the entire chromosome containing the TcNTR gene, a mechanism that does not result in deleterious phenotypic consequences (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008; Mejia et al. Reference Mejia, Hall, Taylor, Gomez-Palacio, Wilkinson, Triana-Chavez and Kelly2012). Other routes to acquired resistance include point mutations in TcNTR that lead to inactivation of the protein product by disruption of FMN-binding capacity. The ease with which this can occur has been demonstrated in a report that three distinct TcNTR inactivating mutations arose independently within a single population undergoing benznidazole selection (Mejia et al. Reference Mejia, Hall, Taylor, Gomez-Palacio, Wilkinson, Triana-Chavez and Kelly2012). Although resistance linked to TcNTR is now well characterized, there is also evidence that other mechanisms can act on drug efficacy. For example, resistance to nifurtimox and benznidazole can occur independently, suggesting a non-involvement of TcNTR in these cases (Filardi and Brener, Reference Filardi and Brener1987). Similarly, a recent survey has found a 10-fold variation in benznidazole sensitivity in parasite isolates from a variety of biological and geographical backgrounds, which is not associated with differences in the sequence of the TcNTR genes (Mejia et al. Reference Mejia, Hall, Taylor, Gomez-Palacio, Wilkinson, Triana-Chavez and Kelly2012), although an association with the level of expression cannot be excluded. Identifying other possible mechanisms of resistance must be regarded as a priority for Chagas disease researchers.
T. brucei resistance to nitroheterocyclic pro-drugs, including nifurtimox and the clinical trial candidate fexnidazole, is also easily selected in the laboratory and is not associated with reduced virulence (Sokolova et al. Reference Sokolova, Wyllie, Patterson, Oza, Read and Fairlamb2010). Sensitivity to these drugs is also associated with activation by NTR in T. brucei (Wilkinson et al. Reference Wilkinson, Taylor, Horn, Kelly and Cheeseman2008) and in Leishmania (Wyllie et al. Reference Wyllie, Patterson, Stojanovski, Simeons, Norval, Kime, Read and Fairlamb2012).
INCREASING THROUGHPUT – GENETIC SCREENS
The ‘low throughput’ approaches described above have identified several resistance mechanisms, some of which have subsequently been found to be clinically important (Table 1). However, they are often dependent upon long-term selection of resistant parasites and prior knowledge enabling the identification of suitable candidate proteins. Also, they provide little access to the wider network of factors that influence drug efficacy. These limitations can be overcome by the development of high throughput systems that enable the contribution of every gene product to be simultaneously assessed (see Fig. 2 for a comparison of low and high throughput approaches). New high throughput approaches, as well as improved tools for low throughput analysis, have led to a significant growth in our understanding of drug-trypanosome interactions over recent years (Fig. 3A). This transition has accompanied the generation, release and exploitation of genome sequence data (Berriman et al. Reference Berriman, Ghedin, Hertz-Fowler, Blandin, Renauld, Bartholomeu, Lennard, Caler, Hamlin, Haas, Bohme, Hannick, Aslett, Shallom, Marcello, Hou, Wickstead, Alsmark, Arrowsmith, Atkin, Barron, Bringaud, Brooks, Carrington, Cherevach, Chillingworth, Churcher, Clark, Corton, Cronin, Davies, Doggett, Djikeng, Feldblyum, Field, Fraser, Goodhead, Hance, Harper, Harris, Hauser, Hostetler, Ivens, Jagels, Johnson, Johnson, Jones, Kerhornou, Koo, Larke, Landfear, Larkin, Leech, Line, Lord, Macleod, Mooney, Moule, Martin, Morgan, Mungall, Norbertczak, Ormond, Pai, Peacock, Peterson, Quail, Rabbinowitsch, Rajandream, Reitter, Salzberg, Sanders, Schobel, Sharp, Simmonds, Simpson, Tallon, Turner, Tait, Tivey, Van Aken, Walker, Wanless, Wang, White, White, Whitehead, Woodward, Wortman, Adams, Embley, Gull, Ullu, Barry, Fairlamb, Opperdoes, Barrell, Donelson, Hall, Fraser, Melville and El-Sayed2005; El-Sayed et al. Reference El-Sayed, Myler, Bartholomeu, Nilsson, Aggarwal, Tran, Ghedin, Worthey, Delcher, Blandin, Westenberger, Caler, Cerqueira, Branche, Haas, Anupama, Arner, Aslund, Attipoe, Bontempi, Bringaud, Burton, Cadag, Campbell, Carrington, Crabtree, Darban, Da Silveira, De Jong, Edwards, Englund, Fazelina, Feldblyum, Ferella, Frasch, Gull, Horn, Hou, Huang, Kindlund, Klingbeil, Kluge, Koo, Lacerda, Levin, Lorenzi, Louie, Machado, Mcculloch, Mckenna, Mizuno, Mottram, Nelson, Ochaya, Osoegawa, Pai, Parsons, Pentony, Pettersson, Pop, Ramirez, Rinta, Robertson, Salzberg, Sanchez, Seyler, Sharma, Shetty, Simpson, Sisk, Tammi, Tarleton, Teixeira, Van Aken, Vogt, Ward, Wickstead, Wortman, White, Fraser, Stuart and Andersson2005; Ivens et al. Reference Ivens, Peacock, Worthey, Murphy, Aggarwal, Berriman, Sisk, Rajandream, Adlem, Aert, Anupama, Apostolou, Attipoe, Bason, Bauser, Beck, Beverley, Bianchettin, Borzym, Bothe, Bruschi, Collins, Cadag, Ciarloni, Clayton, Coulson, Cronin, Cruz, Davies, De Gaudenzi, Dobson, Duesterhoeft, Fazelina, Fosker, Frasch, Fraser, Fuchs, Gabel, Goble, Goffeau, Harris, Hertz-Fowler, Hilbert, Horn, Huang, Klages, Knights, Kube, Larke, Litvin, Lord, Louie, Marra, Masuy, Matthews, Michaeli, Mottram, Muller-Auer, Munden, Nelson, Norbertczak, Oliver, O'neil, Pentony, Pohl, Price, Purnelle, Quail, Rabbinowitsch, Reinhardt, Rieger, Rinta, Robben, Robertson, Ruiz, Rutter, Saunders, Schafer, Schein, Schwartz, Seeger, Seyler, Sharp, Shin, Sivam, Squares, Squares, Tosato, Vogt, Volckaert, Wambutt, Warren, Wedler, Woodward, Zhou, Zimmermann, Smith, Blackwell, Stuart, Barrell and Myler2005). During this period, researchers have developed novel approaches to exploiting these data with impressive effect and these developments have already had a tremendous impact on our understanding of drug resistance in trypanosomes. Indeed, new genetic approaches have recently led to the identification of many genes associated with drug action and resistance phenotypes.

Fig. 2. A comparison of low and high throughput approaches to understanding drug resistance in trypanosomes.

Fig. 3. Developments in T. brucei molecular genetics and advances in the understanding of drug resistance in trypanosomes since 1990. (A) The development of the bloodstream form T. brucei RNAi library and the RIT-seq methodology was dependent upon a number of earlier methodological advances. Although concurrent progress was made in dissecting drug resistance mechanisms in trypanosomes, the establishment of a high throughput approach rapidly led to a significant increase in our understanding of both potential resistance mechanisms and the networks of proteins that influence drug efficacy. See text for abbreviations and references. (B) Drug selection of the tetracycline induced RNAi library can rapidly generate a resistant population. The RNAi targets, whose expression confers drug resistance, are sequenced using RNAi plasmid-specific primers (grey bars) on a next generation sequencing platform, such as Illumina, and mapped to the reference genome to identify candidate drug efficacy determinants.
Prior to sequencing of the trypanosome genomes, the power of the classical forward genetic screen was demonstrated when the gene encoding ODC was recovered by complementation of putrescine auxotrophy in odc mutant T. brucei (Sommer et al. Reference Sommer, Hua, Li, Gottesdiener and Wang1996). This feasibility study suggested that cells exhibiting a phenotype of interest could be isolated from a mutagenized population and the mutagenized gene(s) could be recovered by complementation. Despite the promise of identifying novel genes that control important phenotypes, this form of forward genetic screening has not been widely used. In terms of drug resistance (and many other phenotype screens), this can be partly explained by the difficulty associated with complementation of the drug-resistance phenotype; gain-of-function would increase drug sensitivity, which cannot typically be selected for in the context of a complex population. Another challenge for mutagenesis screens is the typically low frequency of loss-of-heterozygocity in trypanosomes, since many phenotypes are likely to be mild when one allele remains intact in these diploid cells.
Transposon mutagenesis in trypanosomatids (Gueiros-Filho and Beverley, Reference Gueiros-Filho and Beverley1997) raised the possibility of loss-of-function and ‘signature-tagged mutagenesis’ in a single step. This approach was validated using a transposon mutagenesis screen for lectin (concanavalin A) resistance in T. brucei, which lead to the identification of ALG12, a gene involved in N-linked oligosaccharide synthesis (Leal et al. Reference Leal, Acosta-Serrano, Morris and Cross2004). However, as above, the frequency of loss-of-heterozygosity may also represent a limiting factor for these screens.
RNA interference
Potent and specific genetic interference by double-stranded RNA (dsRNA), or RNA interference (RNAi), was first reported in the nematode, Caenorhabditis elegans (Fire et al. Reference Fire, Xu, Montgomery, Kostas, Driver and Mello1998). In the same year, dsRNA was shown to induce mRNA degradation in T. brucei (Ngo et al. Reference Ngo, Tschudi, Gull and Ullu1998) and RNAi has subsequently had a huge impact on genetic studies in African trypanosomes. The genes encoding the RNAi machinery were found to be degenerate or absent in the T. cruzi and Leishmania major genomes, but L. braziliensis retains the machinery for RNAi, indicating that RNAi can be used as a genetic tool in some Leishmania species (Lye et al. Reference Lye, Owens, Shi, Murta, Vieira, Turco, Tschudi, Ullu and Beverley2010).
Several technical innovations and developments have helped to facilitate the wider use of RNAi technology for functional genomics in trypanosomes (Fig. 3A). One of the early innovations was the development of dsRNA inducible expression vectors with head-to-head or opposing promoters (LaCount et al. Reference Lacount, Bruse, Hill and Donelson2000; Wang et al. Reference Wang, Morris, Drew and Englund2000); tools for inducible expression were developed previously (Wirtz and Clayton, Reference Wirtz and Clayton1995; Wirtz et al. Reference Wirtz, Leal, Ochatt and Cross1999). This allowed the rapid assembly of multiple RNAi-targeting vectors, and several hundred T. brucei genes have now been analysed individually using such vectors, including the full complement of RNA polymerase II transcribed genes on chromosome 1 (Subramaniam et al. Reference Subramaniam, Veazey, Redmond, Hayes-Sinclair, Chambers, Carrington, Gull, Matthews, Horn and Field2006).
RNA interference libraries
A particularly important innovation, made possible by the string of technical advances that went before (Fig. 3A), was the development of the T. brucei RNAi library. In fact, the first RNAi library screen in T. brucei was the first RNAi screen carried out in any organism (Morris et al. Reference Morris, Wang, Drew and Englund2002). In this case, concanavalin-A resistance was linked to changes in glycolysis that affected cell-surface protein glycosylation status in procyclic, insect stage cells. This RNAi library has also subsequently been used to select for tubercidin-resistant cells, revealing hexose transporter knockdown and inhibition of glycolysis by this drug (Drew et al. Reference Drew, Morris, Wang, Wells, Sanchez, Landfear and Englund2003). The library has also been used to generate large numbers of individual RNAi strains which have been screened for a variety of phenotypes; see Zhao et al. (Reference Zhao, Lindsay, Roy Chowdhury, Robinson and Englund2008) for example. The potential for further drug-resistance screening was clearly demonstrated at this point in time, but there were some outstanding technical issues. One particularly prominent challenge was to transfer RNAi library screening from the insect-stage of T. brucei to the developmentally distinct and pathogenic bloodstream-form parasite.
One difficulty, which makes comparison among some T. brucei RNAi strains challenging, and may also be responsible for a number of ‘false negative’ or even ‘false positive’ assignments in terms of RNAi-associated phenotypes, is a phenomenon known as position effect. This comes about because dsRNA expression vectors are typically integrated at one of several related chromosomal loci, which support reproducible, but highly variable expression levels. One solution to this problem was to modify a single, expression-validated integration site (a non-transcribed ribosomal spacer) such that all expression vectors are specifically targeted to that site by homologous recombination (Alsford et al. Reference Alsford and Horn2005b). Another challenge was low transfection efficiency in bloodstream-form T. brucei, the stage where drug-resistance studies would typically be more informative. This hurdle was overcome by priming a chromosomal site for homologous recombination by meganuclease cleavage (Glover and Horn, Reference Glover and Horn2009), or by using an improved transfection protocol and pooling many transformed populations (Schumann Burkard et al. Reference Schumann Burkard, Jutzi and Roditi2011). Both approaches can now be used to generate genome-scale RNAi libraries, with the former approach also minimising the position effects described above. Thus, it is now possible to assemble bloodstream-form T. brucei RNAi libraries with approximately 10× genome coverage.
Proof-of-principle, in terms of screening for drug action and resistance mechanisms relevant to the bloodstream life-cycle stage, emerged in 2011. Drug transporters, AT1 and AAT6, and a drug activator, NTR, identified previously by other means (see above), were rapidly identified in RNAi library screens for melarsoprol, eflornithine, and nifurtimox-benznidazole resistance, respectively (Baker et al. Reference Baker, Alsford and Horn2011; Schumann Burkard et al. Reference Schumann Burkard, Jutzi and Roditi2011). Drugs were no longer taken up by these cells or no longer activated once inside, due to the loss-of-function defects associated with inducible RNAi. Another challenge, in terms of whole-genome screening, was then to develop outputs that report phenotypes associated with multiple genes. An early feasibility study showed that it would be possible to report relative representations of large numbers of RNAi clones within complex populations, using a slot-blot or microarray read-out format, for example (Alsford et al. Reference Alsford, Glover and Horn2005a). However, DNA sequencing technology has now become the method of choice for generating complex genetic read-outs and this has been applied to genome-scale libraries using an approach called RNA interference target sequencing, or RIT-seq (Alsford et al. Reference Alsford, Kawahara, Glover and Horn2011). RIT-seq analysis, following RNAi screens for eflornithine, nifurtimox, melarsoprol, pentamidine and suramin resistance (Fig. 3B), revealed more than fifty new genes linked to the uptake or action of these drugs (Alsford et al. Reference Alsford, Turner, Obado, Sanchez-Flores, Glover, Berriman, Hertz-Fowler and Horn2012), including aquaglyceroporin 2, the loss of which leads to melarsoprol-pentamidine cross-resistance (Baker et al. Reference Baker, Glover, Munday, Aguinaga Andres, Barrett, De Koning and Horn2012). This approach can also be used to gain insights into the uptake and intracellular transit of drugs used against related parasites, such as T. cruzi and Leishmania, which while similar to T. brucei are genetically less tractable (Kolev et al. Reference Kolev, Tschudi and Ullu2011). Indeed, the RNAi screen using nifurtimox implicated several factors, in addition to NTR (Alsford et al. Reference Alsford, Turner, Obado, Sanchez-Flores, Glover, Berriman, Hertz-Fowler and Horn2012). More recently, initial RNAi screens using the anti-leishmanials, sodium stibogluconate, amphotericin-B, miltefosine and paromomycin, have identified several proteins already known to interact with these drugs, as well as a number of additional candidate transporters (Alsford, unpublished results).
FUTURE PROSPECTS
We have advanced from single gene analysis to high-throughput, genome-scale screens in a relatively short time. These developments, driven by the availability of genome sequence data, should facilitate further progress. For example, a genome-scale gain-of-function screening approach could now be developed for trypanosomatids. This would involve the generation of an over-expression library, which could be amenable to the same phenotype screens applied to the RNAi loss-of-function libraries, including drug-resistance screens. In T. cruzi, episomal expression (Kelly et al. Reference Kelly, Das and Tomas1992) and cosmid vectors (Kelly et al. Reference Kelly, Ward, Miles and Kendall1994) offer an approach to generating such libraries. The availability of both loss-of-function and gain-of-function libraries for parallel screening could provide further insight into drug-resistance mechanisms, revealing, for example, efflux channels and even the drug-targets themselves. This approach could be applied to new drugs as they advance through development, providing useful information to facilitate the development of new and more effective compounds and combination therapies.
FINANCIAL SUPPORT
Work in the authors’ laboratories is supported by the Wellcome Trust (S. A., Institutional Strategic Support Fellowship; J. M. K., grant number 092573; D. H., grant number 093010/Z/10/Z) and the Medical Research Council (S. A., grant number MR/K011987/1; D. H., grant number MR/K000500/1). N. B. was also supported by a Bloomsbury Colleges doctoral studentship.





 Open access
Open access