


LEARNING OBJECTIVES
After reading this article you will be able to:
• discuss the immunology of acute and chronic stress exposure
• describe the impacts of inflammation on the brain and behaviour in the context of stress-related psychopathology
• consider the clinical implications of the stress of immunology.
Exposure to a vast array of stressors is pervasive throughout our modern-day society and contributes significantly to the risk for adverse behavioural outcomes, including depression and anxiety (McEwen Reference McEwen2008). One critical player in the response to stress and its impact on health is the immune system, which includes both innate and adaptive immune responses. Of special relevance is that the context (e.g. acute versus chronic) of stress exposure can significantly influence how the organism and the immune system respond to threat. Although acute activation of the immune system in response to threat is homeostatically regulated by neuroendocrine mechanisms, chronic activation of the immune system arising from persistent stress exposure can contribute to an allostatic load with an inflammatory diathesis that has been implicated in the pathophysiology of mood and anxiety disorders (McEwen Reference McEwen2008).
Homeostatic regulation of immune activation in response to acute stress exposure
Engagement of the sympathetic nervous system
In the context of acute stressor exposure, rapid engagement of the sympathetic nervous system (SNS) results in the activation of cells mediating the innate and adaptive immune response via efferent projections from the SNS to the bone marrow and lymphoid tissues to prepare the body for injury and wound repair that may result from a threat (Dhabhar Reference Dhabhar, Malarkey and Neri2012). The innate immune response functions quickly (within minutes to hours) to provide organismal defence against pathogens and/or tissue damage or destruction from wounding. This natural immunity is mediated by an array of leukocytes, including granulocytes (neutrophils, eosinophils, basophils and mast cells), monocytes/macrophages and natural killer (NK) cells, which produce inflammation (e.g. cytokines and reactive oxygen species) and engage in phagocytosis to destroy and dispose of the pathogens respectively and initiate the wound healing process. Innate immunity is fast-acting on threat exposure, whereas acquired immunity requires days to generate response to specific pathogens. Cells mediating acquired immunity include different classes of lymphocyte that express antigen-specific receptor sites on their surfaces. The release of adrenaline and noradrenaline from the sympathetic–adrenal–medullary axis on threat exposure activates monocytes/macrophages and lymphocytes via beta-adrenergic receptors to induce the innate and specific immune responses (Kenney Reference Kenney and Ganta2014) respectively.
Leukocyte trafficking of immune cells and upregulation of pro-inflammatory gene expression
Across vertebrate species, including humans, on exposure to an acute stressor, SNS signalling via adrenaline and noradrenaline induces rapid alterations in the absolute numbers and the proportion of leukocytes in circulation that function to traffic immune cells to sites of wounds (Herbert Reference Herbert and Cohen1993). This occurs in tandem with a redistribution of leukocytes within compartments critical for immune system function, as there is an initial increase in lymphocytes and monocytes in the blood that is subsequently followed by a decrease as these cells enter organ compartments, such as the skin, lungs and lymph nodes, that may be a site of wounding and/or infiltration by pathogens (Dhabhar Reference Dhabhar, Malarkey and Neri2012). For instance, acute stress exposure (e.g. physical restraint) in mice results in a more robust increase in the infiltration of leukocytes, including neutrophils, macrophages and NK and T cells, at the site of surgery or wounding (Viswanathan Reference Viswanathan and Dhabhar2005a). A concomitant upregulation in pro-inflammatory gene expression, including tumour necrosis factor (TNF), interferon-gamma (IFN-γ), and interleukins 1-beta (IL-1β) and 6 (IL-6), occurs at the site of this acute stress-induced redistribution of immune cells (Viswanathan Reference Viswanathan, Daugherty and Dhabhar2005b).
The ability of acute stress exposure to induce changes in gene expression is mediated by the activity of nuclear factor kappa B (NF-κB), a redox-sensitive transcription factor whose activity increases pro-inflammatory cytokine secretion from mononuclear cells. More specifically, translational studies show that increases in noradrenaline following acute psychosocial stress (e.g. the Trier Social Stress Test, TSST) and immobilisation stress (e.g. restraint) exposure in humans and mice respectively activates NF-κB to induce IL-6 release (Bierhaus Reference Bierhaus, Wolf and Andrassy2003). In vitro and in vivo studies also show that pharmacological blockade of adrenergic signalling via α1-adrenergic antagonists blocks this stress-induced NF-κB activity (Bierhaus Reference Bierhaus, Wolf and Andrassy2003). It is important to note, however, that the ability of adrenaline to induce pro- or anti-inflammatory cascades within the innate immune system is dependent on cell-specific expression of different β-adrenergic receptor subtypes (Kenney Reference Kenney and Ganta2014).
Glucocorticoid release
Adrenaline and noradrenaline release on threat-induced activation of the sympathetic–adrenal–medullary axis occurs in tandem with aldosterone release that acts via mineralocorticoid receptors to decrease neutrophils, helper T cells and NK cells (Miller Reference Miller, Spencer and Hassett1994). Parallel threat-induced activation of the hypothalamic–pituitary–adrenal (HPA) axis results in de novo cortisol synthesis and release from the adrenal cortex that acts via glucocorticoid receptors to affect immune cell distribution and activity (Miller Reference Miller, Spencer and Hassett1994).
Low concentrations of corticosterone in rodents have been shown to enhance acute stress-induced redistribution of T cells and delayed-type hypersensitivity (DTH) of the skin, while also leading to trafficking of immune cells to the brain (meninges) in association with decreased anxiety-like behaviour (Lewitus Reference Lewitus, Cohen and Schwartz2008). In contrast, high doses and chronic doses of corticosterone or the synthetic glucocorticoid dexamethasone suppress DHT (Dhabhar Reference Dhabhar and McEwen1999) and increase anxiety- and depression-like behaviours in laboratory animals. This bimodal, or biphasic, response of the immune system to acute stress-induced release of glucocorticoids is dependent on negative feedback mechanisms at the level of glucocorticoid receptors to inhibit NF-κB and the downstream release of pro-inflammatory cytokines, and acts to restore homeostasis (McEwen Reference McEwen and Wingfield2010).
Summary
Taken together, existing data indicate that acute stress exposure enhances innate and acquired immunity to increase the chances of organismal survival in the face of potential wounding and pathogen entry, whereas chronic exposure to stress may have more detrimental effects.
Chronic stress-induced allostasis facilitates increased systemic inflammation
Glucocorticoid resistance
Under conditions wherein organisms are exposed to chronic (e.g. unrelenting, constant) stressors, glucocorticoid negative feedback inhibition of immune activation is impaired in a manner that drives allostasis (i.e. maintenance of organismal stability by altering physiological properties to counteract threats) (McEwen Reference McEwen and Wingfield2010) and facilitates increased levels of systemic inflammation. Chronic stress exposure results in diminished glucocorticoid negative feedback of the HPA axis arising from glucocorticoid resistance (Cohen Reference Cohen, Janicki-Deverts and Doyle2012). Glucocorticoid resistance is believed to be due in part to inhibitory effects of cytokines on glucocorticoid receptor function, as well as stress-induced epigenetic modifications of molecules that regulate the glucocorticoid receptor, including the chaperone protein FKBP5 (Zannas Reference Zannas, Jia and Hafner2019). Consequences of this glucocorticoid resistance include hypercortisolaemia and increased activation of the immune system that can result in heightened pro-inflammatory cytokine and increased risk for individuals to become sick on exposure to pathogens (Cohen Reference Cohen, Janicki-Deverts and Doyle2012).
Alterations in immune gene expression
Translational work in female rhesus macaques, in which the chronic psychosocial stress exposure associated with social subordination (e.g. constant harassment from higher-ranking animals) can be manipulated via social rank rearrangements, shows that low social status causally alters immune gene expression profiles of NK cells, helper T cells, B cells and cytotoxic T cells towards expression profiles that denote increased lymphocyte proliferation, heightened innate immune responses and augmented cytokine responses (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016). These social stress effects on pro-inflammatory gene expression at rest (e.g. in the absence of pathogen exposure) are most potently seen in NK and helper T cells, and are exacerbated on in vitro stimulation with lipopolysaccharide (LPS) (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016), a component of gram-negative bacteria that is used commonly to invoke a strong inflammatory response by binding toll-like receptor 4 (TLR4) on monocytes. More specifically, LPS stimulation in subordinate, chronically stressed monkeys results in the enrichment of genes associated with response to bacterial infection, including the inflammatory response and cytokine production (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016), and lower expression of genes involved in the antiviral response and type I interferon signalling (Sanz Reference Sanz, Maurizio and Snyder-Mackler2019). Of note, this stress-related increase in expression of inflammatory genes and decreased antiviral genes (labelled the conserved transcriptional response to adversity) is believed to be related to chronic sympathetic nervous system activation and has been found in the context of a variety of chronic stressors in humans, including low socioeconomic status (Knight Reference Knight, Rizzo and Logan2016).
Genes upregulated by LPS and more highly expressed in subordinate female macaques include members of the NF-κB transcription factor complex, including NFKBID, NFKBIZ and NFKB1, as well as the STAT3 and STAT5A transcription factors that are involved in pro-inflammatory cytokine response (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016). This LPS-induced increase in NF-κB activity in chronically stressed monkeys is due to the polarisation of the TLR4 signalling cascade towards the inflammatory MyD88-dependent pathway and away from the antiviral TRIF-dependent pathway that is favoured in more dominant monkeys (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016; Sanz Reference Sanz, Maurizio and Snyder-Mackler2019). Interestingly, social subordination also drives an exaggerated expression of NF-κB and interferon-associated genes on challenge with a viral mimic that can also be pro-inflammatory in nature (Sanz Reference Sanz, Maurizio and Snyder-Mackler2019). Critically, the increase in pro-inflammatory response on LPS stimulation is mediated by diminished glucocorticoid sensitivity in low-ranking animals (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016).
Changes in chromatin structure
The alterations in immune cell gene expression described above due to chronic psychosocial stress exposure in female rhesus macaques are associated with changes in chromatin structure and thus in DNA accessibility to glucocorticoids (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2016). More specifically, low-ranking females present chromatin landscapes that are more accessible for NF-κB transcription factor binding sites, whereas high-ranking females show more accessible binding sites for AP-1, the glucocorticoid receptor cofactor that is involved in anti-inflammatory responses and inhibition of NF-κB (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2019). Importantly, in vitro dexamethasone administration also results in the enrichment of transcription factor binding sites for AP-1, suggesting that glucocorticoid resistance resulting from chronic subordination stress alters the dynamics of glucocorticoid-mediated gene expression in immune cells and chromatic accessibility to drive systemic inflammation (Snyder-Mackler Reference Snyder-Mackler, Sanz and Kohn2019).
Summary
Taken together, these data underscore the mechanisms by which maladaptive allostatic consequences of chronic stress exposure can drive a pro-inflammatory state that increases risk for adverse health outcomes, including stress-related psychopathology (McEwen Reference McEwen2008).
Increased inflammation in individuals with stress-related psychopathology
Systemic inflammation is associated with stress-related psychopathology (McEwen Reference McEwen2008). Systematic reviews and meta-analyses of available data on the relationship between inflammation and depression and fear and anxiety disorders support the notion that these stress-related conditions are associated with increased systemic inflammation as assessed by circulating concentrations of C-reactive protein (CRP) and cytokines (Table 1). Meta-analytic results indicate that glucocorticoid resistance is an important component of this increased inflammation in patients with depression (Perrin Reference Perrin, Horowitz and Roelofs2019).
TABLE 1 Summary of systematic reviews and meta-analyses on the relationship between inflammation and stress-related psychopathology

CRP, C-reactive protein; TNF, tumour necrosis factor; IL, interleukin; IFN, interferon; n.a., not assessed; –, null finding; ↑, significantly higher in cases compared with healthy controls; s, soluble; PTSD, post-traumatic stress disorder.
It is important to note that, although most work to date focuses on peripheral blood concentrations of inflammatory biomarkers in stress-related psychiatric conditions (Table 1) (Modabbernia Reference Modabbernia, Taslimi and Brietzke2013; Dargel Reference Dargel, Godin and Kapczinski2015; Quagliato Reference Quagliato and Nardi2018; Renna Reference Renna, O'Toole and Spaeth2018; Cosco Reference Cosco, Pillinger and Emam2019; Costello Reference Costello, Gould and Abrol2019; Osimo Reference Osimo, Pillinger and Rodriguez2020; Yang Reference Yang and Jiang2020), heightened inflammation is also seen in the brain. More specifically, a systematic review and meta-analysis shows that cerebral spinal fluid (CSF) concentrations of IL-6 and TNF are increased in individuals with depression (Enache Reference Enache, Pariante and Mondelli2019). Increased microglia activation, the central mediators of the immune system, as assessed by positron emission tomography (PET) neuroimaging, and greater expression of TNF and TLR4 in post-mortem brain tissue have also been described in individuals with depression (Enache Reference Enache, Pariante and Mondelli2019).
Although these large-scale and reproducible data highlight the association between inflammation and stress-related psychiatric disorders, the cross-sectional nature of the majority of the studies limits our ability to determine the cause and effect relationship between stress-induced inflammation and behaviour. Nevertheless, translational studies in human and pre-clinical models clearly show that peripheral and central inflammation can have a direct impact on brain function to drive psychiatric symptoms and that blocking inflammation can reduce symptoms of depression and anxiety in patients with increased inflammation.
Mechanisms by which inflammation contributes to stress-related symptoms
The notion that stress-induced inflammation can induce affective symptomology was first highlighted by the finding that use of the inflammatory cytokine interferon-alpha to treat infectious diseases and cancer induced depressive symptoms (Raison Reference Raison, Demetrashvili and Capuron2005). Since then, a significant body of literature has emerged that describes the causal effects of acute and chronic inflammatory stimuli on the emergence of affective symptoms. For example, administration of typhoid vaccination induces depressed mood, anhedonia and fatigue (Harrison Reference Harrison, Brydon and Walker2009). Neuroimaging studies have further shown that these inflammatory stimuli, as well as endogenous inflammation in patients with depression, can alter the functional connectivity and activation of brain regions implicated in the pathophysiology of stress-related psychopathology, including the prefrontal cortex, striatum, dorsal anterior cingulate cortex and amygdala (Harrison Reference Harrison, Brydon and Walker2009). These studies indicate that inflammation decreases functional connectivity between the prefrontal cortex and striatum in a manner that predicts reward deficits, anhedonia and psychomotor slowing.
Peripheral inflammatory cytokines crossing the blood–brain barrier
Cytokines released in the periphery in response to stress exposure can affect the brain by passing through leaky regions of the blood–brain barrier, being actively transported across it, activating endothelial and perivascular macrophages lining the brain to release their own cytokines into the brain parenchyma, and activating cytokine receptors on the vagus nerve and other peripheral afferent nerves to signal the brain (Haroon Reference Haroon, Raison and Miller2012). Peripheral cytokines released in response to stress exposure can also recruit activated monocytes and macrophages from the blood into the brain, wherein they produce their own cytokines and activate microglia, which themselves can release cytokines locally in the brain (D'Mello Reference D'Mello, Le and Swain2009). Finally, more recent data indicate that stress-induced activation of NF-κB and TNF signalling pathways in endothelial cells in the nucleus accumbens can lead to a local reduction in the integrity of the blood–brain barrier (Menard Reference Menard, Pfau and Hodes2017), allowing direct access of inflammatory cytokines to this brain region and, ultimately, depression-like behaviour.
Once in the brain, cytokines can influence behaviour via their ability to alter the metabolism of neurotransmitters, including monoamines and glutamate. These effects of central cytokines on neurotransmitters are mediated through effects on neurotransmitter synthesis, release and reuptake, leading to decreased monoamine availability and increased extrasynaptic glutamate, which can be excitotoxic (Haroon Reference Haroon, Raison and Miller2012). In addition, increased activation of indoleamine 2,3-dioxygenase (IDO), the enzyme that acts to convert tryptophan into kynurenine, leads to greater levels of kynurenine. This increased kynurenine is then broken down into quinolinic acid, an N-methyl-d-aspartate (NMDA) receptor agonist, which can further contribute to glutamate excitotoxicity and oxidative stress (Haroon Reference Haroon, Raison and Miller2012). Cytokine-induced alterations in IDO/kynurenine can decrease serotonin and dopamine levels, as well as increase glutamate levels, which have been linked to increased stress-related symptoms, including depressed mood, anhedonia and psychomotor slowing (Haroon Reference Haroon, Raison and Miller2012).
Clinical considerations and implications regarding the immunology of stress
Under conditions of chronic exposure to stressors, the emergence of a pro-inflammatory allostatic state can contribute to psychiatric symptoms across depression and anxiety disorders via site-specific cytokine actions on neurotransmitter systems in brain regions underlying emotion regulation and affect (Fig. 1). Accordingly, interventions targeting the immune system and its downstream effects on the brain for the treatment of depression and other psychiatric disorders have been of great interest. A number of strategies have been employed, including blocking inflammation itself through pharmacological or behavioural means or attempting to reverse the downstream effects of inflammation on neurotransmitter systems.
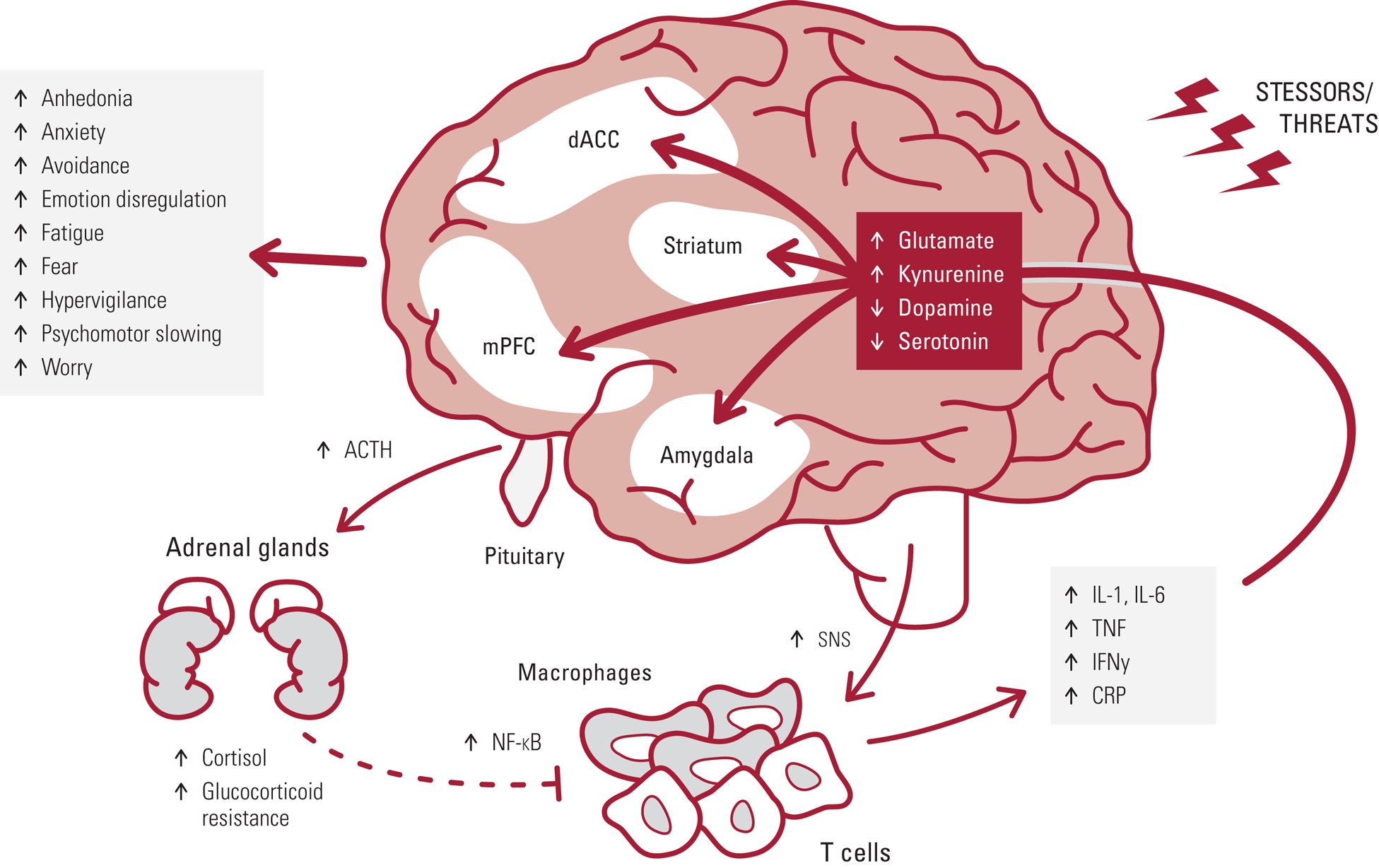
FIG 1 Exposure to chronic stressors and threats drives adrenocorticotropic hormone (ACTH) and cortisol release, as well as increased activity of the sympathetic nervous system (SNS). SNS activation of NF-κB activity in immune cells increases expression of pro-inflammatory cytokines (e.g. IL-1,IL-6, TNF, IFN-γ) and CRP. Glucocorticoid resistance develops wherein cortisol does not as effectivity inhibit NF-κB activity, thus creating a pro-inflammatory allostatic state that can contribute to psychiatric symptoms via cytokine actions on glutamate, kynurenine, dopamine and serotonin systems in brain regions underlying emotion regulation and affect, including the striatum, dorsal anterior cingulate (dACC), medial prefrontal cortex (mPFC) and amygdala.
Blocking inflammation using cytokine antagonists
Probably the most convincing data that blocking inflammation can reduce depressive symptoms comes from studies using cytokine antagonists in people with autoimmune and inflammatory disorders, albeit the impact of these drugs on the underlying disease complicate interpretation of these findings. Meta-analyses of other medications that putatively target the impact of inflammation on the brain, including COX-2 inhibitors, aspirin and minocycline (a tetracycline antibiotic that decreases microglial activation), have revealed some evidence of effectiveness in otherwise healthy depressed individuals. However, the off-target effects of these medications and the fact that increased inflammation occurs in only about one-third of depressed patients leaves some level of doubt regarding the specificity of findings relative to inflammation (Osimo Reference Osimo, Baxter and Lewis2019; Bavaresco Reference Bavaresco, Uggioni and Ferraz2020). Only a handful of studies have used anti-cytokine therapies in depression, and the results suggest that baseline inflammation (as reflected by CRP) is an important predictor of response, and symptoms that seem most responsive relate to anhedonia, psychomotor retardation and anxiety (Bavaresco Reference Bavaresco, Uggioni and Ferraz2020).
The notion that baseline levels of inflammation may be an important consideration for pharmacological treatment extends past the use of anti-inflammatory agents, as translational studies have shown that greater inflammation in depression is also associated with resistance to conventional antidepressant treatments (Chamberlain Reference Chamberlain, Cavanagh and de Boer2019). The variability in the inflammatory profiles of individuals diagnosed with major depression is highlighted by a recent systematic review and meta-analysis reporting that approximately a quarter of individuals with major depressive disorder show low-grade inflammations (CRP level >3 mg/L) and approximately half show mildly elevated CRP levels (CRP >1 mg/L) (Osimo Reference Osimo, Baxter and Lewis2019).
SSRIs, cognitive and behavioural interventions, ketamine and l-dopa
Other pharmacological and behavioural interventions shown to be efficacious for the treatment of stress-related psychopathology may be immunomodulatory in nature, and thus could provide benefits through their abilities to attenuate systemic inflammation (Liu Reference Liu, Wei and Strawbridge2020). Therapy with selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine, paroxetine, sertraline, citalopram, escitalopram and fluvoxamine decreases peripheral concentrations of IL-6, IL-1β and TNF (Wang Reference Wang, Wang and Liu2019), although these effects appear to be largely related to treatment response and probably the associated reduction in stress. Mindfulness-based interventions have been shown in meta-analyses to decrease biomarkers of inflammation, including IL-6 and TNF, across depression and anxiety disorders (Sanada Reference Sanada, Montero-Marin and Barcelo-Soler2020), and cognitive–behavioural therapy for the treatment of depression normalises cytokine levels (Dahl Reference Dahl, Ormstad and Aass2016). Finally, drugs such as ketamine (an NMDA antagonist) and levodopa (l-dopa, a precursor of dopamine) that are efficacious for treating depression (Park Reference Park, Falodun and Zarate2019) may be acting by blocking or circumventing the downstream effects of stress-induced cytokines on glutamate or dopamine respectively.
Implications for future research
Although existing data suggest that targeting the immunology of chronic stress may be a valid intervention for stress-related psychopathology, the majority of research to date has taken place in the context of depression. Future translational and clinical research is necessary to better determine the mechanism by which the immune system and inflammation contribute to anxiety disorders, and whether interventions targeting the immune system or its effects on the brain are efficacious in these conditions. Other factors that contribute significantly to individual variability in the immunology of stress exposure and may be important for treatment considerations in stress-related psychopathology include genetics and epigenetics, biological sex and the presence of other sources of inflammation that may interact with stress, such as smoking, diet and comorbid medical conditions, including obesity, metabolic syndrome, diabetes, cardiovascular disease or cancer. It is also important that more long-term studies leveraging these approaches are undertaken to assess whether the efficacy of the treatments are long-lasting, even in conditions where individuals continue to be exposed to chronic stressors. Finally, it is important to understand the maladaptive mental health consequences of the immunology of stress across the lifespan, starting in childhood.
Author contributions
All authors drafted the manuscript and contributed important intellectual content. All authors approved the final version of this manuscript.
Funding
This review was supported in part by the National Institute of Health, grants AG057235 (V.M.), MH115174 (V.M.) and AG062334 (V.M.).
Declaration of interest
None.
ICMJE forms are in the supplementary material, available online at https://doi.org/10.1192/bja.2020.82.
MCQs
Select the single best option for each question stem
1 Current research indicates that the proportion of individuals with major depression who are thought to have significantly elevated peripheral concentrations of CRP (>3 mg/L) is:
a 33%
b 25%
c 50%
d 80%
e 66%.
2 Which does not occur under homeostatic regulation of the immune system on acute stress/threat exposure?
a SNS activation of macrophages and T cells
b glucocorticoid resistance
c glucocorticoid negative feedback inhibition of NF-κB activation
d increase in innate and acquired immunity to facilitate the chances of organismal survival in the face of potential wounding and pathogens
e acute redistribution of immune cells.
3 Decreased sensitivity of glucocorticoid receptors that occurs with chronic stress/threat exposure does not result in:
a upregulation of pro-inflammatory gene expression
b increased activation of microglia in the brain
c increased ability for glucocorticoids to shut down stress-induced activation of the immune system
d dysregulation of central neurotransmitter metabolism and function
e increased symptoms of anhedonia, anxiety and fatigue.
4 Current research indicates that inflammatory insults affect the activity of:
a prefrontal cortex
b amygdala
c anterior cingulate cortex
d striatum
e all of the above.
5 The most recent meta-analyses indicate that the marker(s) of inflammation not consistently elevated in depression is/are:
a C-reactive protein
b IL-6
c TNF
d IFN-γ
e TNF and IL-6.
MCQ answers
1 b 2 b 3 c 4 e 5 d



eLetters
No eLetters have been published for this article.