


No CrossRef data available.
Article contents
Shinarumpite, a new cobalt uranyl sulfate mineral from the Scenic mine, San Juan County, Utah, USA, structurally related to leydetite
Published online by Cambridge University Press: 28 November 2022
Abstract
The new mineral shinarumpite (IMA2021-105), [Co(H2O)6][(UO2)(SO4)2(H2O)]⋅4H2O, was found in the Scenic mine on Fry Mesa, White Canyon district, San Juan County, Utah, USA, where it occurs as a secondary phase on granular quartz matrix in association with gypsum, deliensite, Co-rich rietveldite, scenicite, shumwayite and sulfur. Shinarumpite crystals are transparent, yellow, blades or prisms, up to 1 mm in length. The mineral has white streak, vitreous lustre and is nonfluorescent. It is brittle with irregular, curved fracture. The Mohs hardness is ~2½ and it has a perfect {100} cleavage. The density is 2.58(2) g⋅cm–3. Optically, the mineral is biaxial (–) with α = 1.515(2), β = 1.526(2), γ = 1.529(2) (white light); 2V = 55(1)°; extreme r < v dispersion; orientation: Z = b, X ^ a = 30° in obtuse β; pleochroism: X = very pale yellow, Y = pale yellow, Z = light yellow; X < Y < Z. The Raman spectrum exhibits bands consistent with UO22+, SO42– and O–H. Electron microprobe analysis provided the empirical formula [(Co0.51Ni0.28Fe0.21)Σ1.00(H2O)6][(UO2)(SO4)2(H2O)]⋅4H2O. The five strongest powder X-ray diffraction lines are [dobs Å(I)(hkl)]: 10.37(100)(200), 5.73(43)(111), 5.20(70)(400, 202, 211), 4.70(31)($\bar{3}$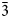 11) and 3.326(30)(213, 021). Shinarumpite is monoclinic, P21/c, a = 21.0549(15), b = 6.8708(5), c = 12.9106(5), β = 96.678(7)°, V = 1885.03(17) Å3 and Z = 4. In the structure of shinarumpite (R1 = 0.0336 for 2623 I > 2σI), linkages of pentagonal bipyramids and tetrahedra form an infinite [(UO2)(SO4)2(H2O)]2– sheet. Isolated Co(H2O)6 octahedra and H2O groups occupy the interlayer region linking the sheets via an extensive system of hydrogen bonds. The structure of shinarumpite is very similar to that of leydetite. Uranyl sulfate structural unit types are discussed with respect to frequency and charge deficiency per anion (CDA).
11) and 3.326(30)(213, 021). Shinarumpite is monoclinic, P21/c, a = 21.0549(15), b = 6.8708(5), c = 12.9106(5), β = 96.678(7)°, V = 1885.03(17) Å3 and Z = 4. In the structure of shinarumpite (R1 = 0.0336 for 2623 I > 2σI), linkages of pentagonal bipyramids and tetrahedra form an infinite [(UO2)(SO4)2(H2O)]2– sheet. Isolated Co(H2O)6 octahedra and H2O groups occupy the interlayer region linking the sheets via an extensive system of hydrogen bonds. The structure of shinarumpite is very similar to that of leydetite. Uranyl sulfate structural unit types are discussed with respect to frequency and charge deficiency per anion (CDA).
Keywords
Information
- Type
- Article
- Information
- Copyright
- Copyright © The Author(s), 2022. Published by Cambridge University Press on behalf of The Mineralogical Society of Great Britain and Ireland.
Footnotes
Associate Editor: Michael Rumsey
References
Bartlett, J.R., Cooney, R.P. (1989) On the determination of uranium-oxygen bond lengths in dioxouranium(VI) compounds by Raman spectroscopy. Journal of Molecular Structure, 193, 295–300.Google Scholar
Burns, P.C. and Nyman, M. (2018) Captivation with encapsulation: a dozen years of exploring uranyl peroxide capsules. Dalton Transactions, 47, 5916–5927.Google Scholar
Čejka, J., Sejkora, J., Mrázek, Z., Urbanec, Z. and Jarchovský, T. (1996): Jáchymovite, (UO2)8(SO4)(OH)14⋅13H2O, a new uranyl mineral from Jáchymov, the Krušné Hory Mts., Czech Republic, and its comparison with uranopilite. Neues Jahrbuch für Mineralogie Abhandlungen, 170, 155–170.Google Scholar
Chenoweth, W.L. (1993) The Geology and Production History of the Uranium Deposits in the White Canyon Mining District, San Juan County, Utah. Utah Geological Survey Miscellaneous Publication, 93-3.Google Scholar
Colmenero, F., Plášil, J., Němec, I. (2020) Uranosphaerite: Crystal structure, hydrogen bonding, mechanics, infrared and Raman spectroscopy and thermodynamics. Journal of Physics and Chemistry of Solids, 141, 109400.Google Scholar
Ferraris, G. and Ivaldi, G. (1988) Bond valence vs bond length in O⋯O hydrogen bonds. Acta Crystallographica Section, B44, 341–344.Google Scholar
Gagné, O.C. and Hawthorne, F.C (2015) Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Crystallographica, B71, 562–578.Google Scholar
Gilbert, G.K. (1875) Report upon the geology of portions of Nevada, Utah, California, and Arizona, examined in the years 1871 and 1872. Pp. 17–187 in: Report on the Geographical and Geological Explorations and Surveys West of the One Hundredth Meridian (Wheeler). U.S. Geological and Geographical Survey, Publication of the Wheeler Survey, Volume 3.Google Scholar
Gunter, M.E., Bandli, B.R., Bloss, F.D., Evans, S.H., Su, S.C., and Weaver, R. (2004) Results from a McCrone spindle stage short course, a new version of EXCALIBR, and how to build a spindle stage. The Microscope, 52, 23–39.Google Scholar
Hawthorne, F.C. (2014) The structure hierarchy hypothesis. Mineralogical Magazine, 78, 957–1027.Google Scholar
Kampf, A.R., Plášil, J., Kasatkin, A.V. and Marty, J. (2015) Bobcookite, NaAl(UO2)2(SO4)4⋅18H2O, and wetherillite, Na2Mg(UO2)2(SO4)4⋅18H2O, two new uranyl sulfate minerals from the Blue Lizard mine, San Juan County, Utah, USA. Mineralogical Magazine, 79, 695–714.Google Scholar
Kampf, A.R., Plášil, J., Kasatkin, A.V., Marty, J. Čejka, J. and Lapčák, Ladislav (2017a) Shumwayite, [(UO2)(SO4)(H2O)2]2⋅H2O, a new uranyl sulfate mineral from Red Canyon, San Juan County, Utah, USA. Mineralogical Magazine, 81, 273–285.Google Scholar
Kampf, A.R., Plášil, J., Čejka, J., Marty, J., Škoda, R. and Lapčák, L. (2017b) Alwilkinsite-(Y), a new rare-earth uranyl sulfate mineral from the Blue Lizard mine, San Juan County, Utah, USA. Mineralogical Magazine, 81, 895–907.Google Scholar
Kampf, A.R., Plášil, J., Olds, T.A., Ma, C. and Marty, J. (2022a) Shinarumpite, IMA 2021-105. CNMNC Newsletter 66. Mineralogical Magazine, 86, https://doi.org/10.1180/mgm.2022.33Google Scholar
Kampf, A.R., Plášil, J., Olds, T.A., Ma, C. and Marty, J. (2022b) Scenicite, a new uranyl-sulfate mineral from the White Canyon district, San Juan County, Utah, USA. Mineralogical Magazine, 86, 743–748, https://doi.org/10.1180/mgm.2022.53Google Scholar
Libowitzky, E. (1999) Correlation of O–H stretching frequencies and O–H⋅⋅⋅O hydrogen bond lengths in minerals. Monatshefte für Chemie, 130, 1047–1059.Google Scholar
Lussier, A.J., Lopez, R.A. and Burns, P.C. (2016) A revised and expanded structure hierarchy of natural and synthetic hexavalent uranium compounds. The Canadian Mineralogist, 54, 177–283.Google Scholar
Mandarino, J.A. (1976) The Gladstone-Dale relationship – Part 1: derivation of new constants. The Canadian Mineralogist, 14, 498–502.Google Scholar
Mandarino, J.A. (2007) The Gladstone–Dale compatibility of minerals and its use in selecting mineral species for further study. The Canadian Mineralogist, 45, 1307–1324.Google Scholar
Nováček, R. (1935) Study of some secondary uranium minerals. Věstník Královské České Společnosti Nauk, 7, 1–16.Google Scholar
Olds, T.A., Plášil, J., Kampf, A.R., Simonetti, A., Sadergaski, L.R., Chen, Y.-S. and Burns, P.C. (2017) Ewingite: Earth's most complex mineral. Journal of Geology, 45, 1007–1010.Google Scholar
Plášil, J., Buixaderas, E., Čejka, J., Sejkora, J., Jehlička, J. and Novák, M. (2010) Raman spectroscopic study of the uranyl sulphate mineral zippeite: low wavenumber and U–O stretching regions. Analytical and Bioanalytical Chemistry, 397, 2703–2715.Google Scholar
Plášil, J., Kasatkin, A.V., Škoda, R., Novák, M., Kallistová, A., Dušek, M., Skála, R., Fejfarová, K., Čejka, J., Meisser, N., Goethals, H., Machovič, V. and Lapčák., L. (2013) Leydetite, Fe(UO2)(SO4)2(H2O)11, a new uranyl sulfate mineral from Mas d'Alary, Lodève, France. Mineralogical Magazine, 77, 429–441.Google Scholar
Plášil, J., Kampf, A.R., Ma, Ch. and Desor, J. (2022) Oldsite, K2Fe2+[(UO2)(SO4)2]2(H2O)8, a new uranyl sulfate mineral from Utah, USA: its description and implications to the formation and occurrences of uranyl sulfate minerals. Mineralogical Magazine, https://doi.org/10.1180/mgm.2022.106Google Scholar
Schindler, M. and Hawthorne, F.C. (2001) A bond-valence approach to the structure, chemistry, and paragenesis of hydroxyl-hydrated oxysalt minerals. I. Theory. The Canadian Mineralogist, 39, 1225–1242.Google Scholar
Schindler, M. and Hawthorne, F.C. (2008) The stereochemistry and chemical composition of interstitial complexes in uranyl-oxysalt minerals. The Canadian Mineralogist, 46, 467–501.Google Scholar
Sheldrick, G.M. (2015a) SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallographica, A71, 3–8.Google Scholar
Sheldrick, G.M. (2015b) Crystal structure refinement with SHELXL. Acta Crystallographica, C71, 3–8.Google Scholar
Vlček, V., Čejka, J., Císařová, I., Goliáš, V. and Plášil, J. (2009) Crystal structure of UO2SO4⋅2.5H2O: Full anisotropic refinement and vibration characterization. Journal of Molecular Structure, 936, 75–79.Google Scholar

Kampf et al. supplementary material
Kampf et al. supplementary material

File
271.9 KB