Introduction
The essential liver endocrine and exocrine functions require a precise spatial arrangement of the repeated hepatic lobule consisting of the central vein, portal vein, hepatic artery, intrahepatic bile duct system, and hepatocyte zonation. This allows (1) blood to be carried through the liver parenchyma and sampled by all hepatocytes, (2) hepatocytes to uptake metabolites and toxins from the blood for metabolizing and detoxification from their basal sinusoidal side, and (3) hepatocytes to produce and secrete bile from their apical canalicular side to be carried out of the liver through the biliary (i.e., intrahepatic bile duct) system composed of biliary epithelial cells (i.e., cholangiocytes).
This chapter describes the process of liver development (i.e., hepatogenesis), defining the molecular factors required for regulating induction of liver progenitors (i.e., hepatoblasts), hepatoblast differentiation into the two epithelial cell types of the liver (i.e., hepatocytes and cholangiocytes), and setting up the initial architecture that is critical for the liver to perform its functions. Understanding these processes and its molecular control provides insight into cellular origin, developmental abnormalities potentially contributing to liver disease, and liver regeneration.
Overview of Hepatogenesis
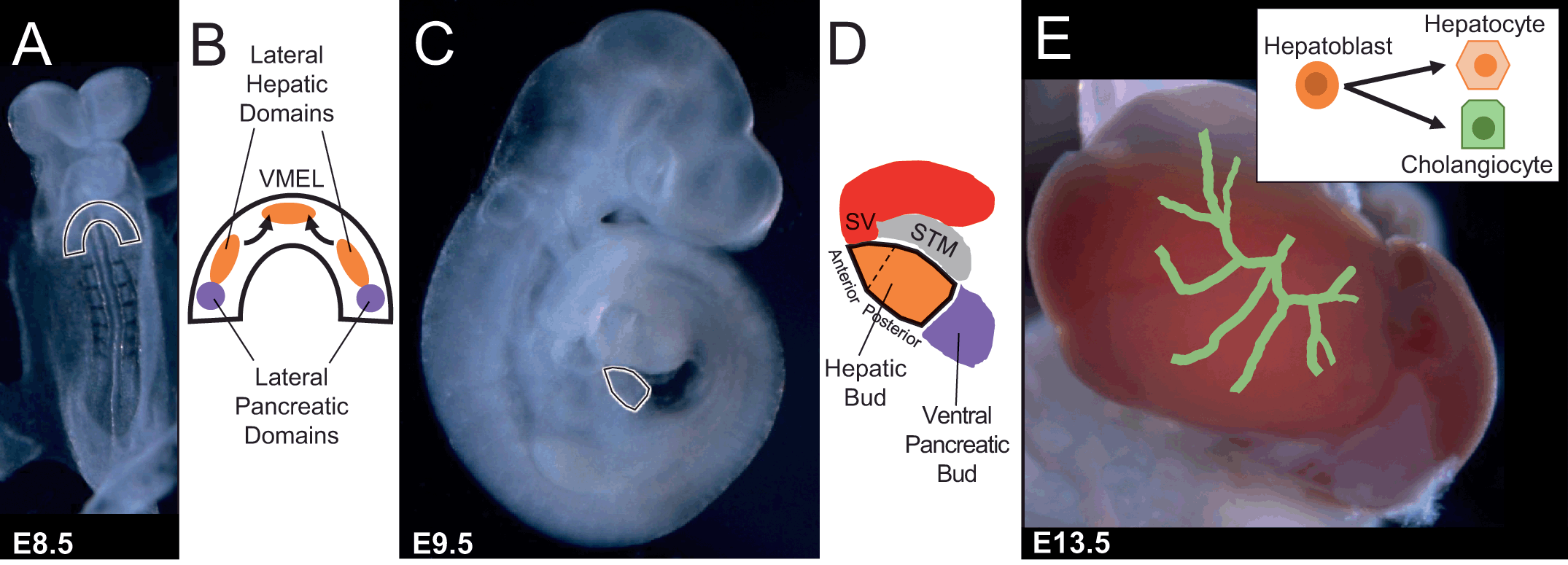
In both mouse and human, the liver originates from the definitive endoderm. There are three germ or primary cell layers formed during early embryonic development consisting of the ectoderm (outer), mesoderm (middle), and endoderm (inner). The definitive endoderm is the embryonic germ layer formed during gastrulation, emerging as an epithelial sheet that lines the ventral surface of the early embryo by approximately human day 16 of gestation and mouse embryonic day (E) 7.5. Ultimately the definitive endoderm produces the digestive, respiratory, thymus, and thyroid organs. Using cell labeling approaches to fate map cellular contribution of definitive endoderm cells as early as mouse E7.5 has demonstrated that it is the ventral foregut endoderm that gives rise to the lung, liver and ventral pancreatic diverticula [Reference El Sebae1]. Refined cell fate mapping reveals that mouse hepatic morphogenesis begins with thickening of the ventral midline of the endoderm lip (VMEL) and two more posterior bilaterally symmetrical lateral domains (Figure 1.1). All three of these domains coalesce to form the hepatic diverticulum [Reference Tremblay and Zaret2]. The cells of the VMEL mainly contribute to the anterior portion and a narrow stripe of cells within the ventral midline of the hepatic bud [Reference Tremblay and Zaret2]. In contrast, the two lateral domains give rise to the majority of the middle and posterior parts of the hepatic bud [Reference Angelo and Tremblay3].
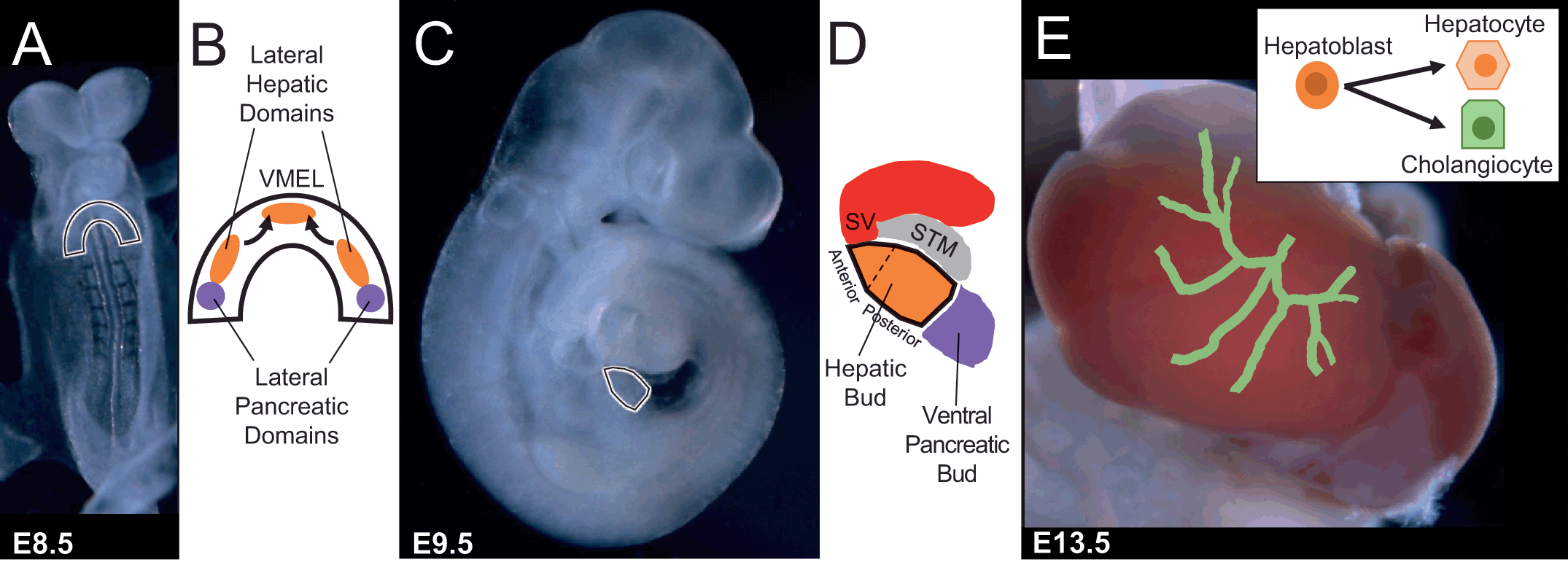
Figure 1.1 Overview of hepatogenesis. (A) E8.5 mouse embryo with ventral foregut endoderm outlined prior to coalescence and formation of the hepatic bud. (B) Schematic of ventral foregut endoderm paired lateral domains and ventral midline endodermal lip (VMEL) from which liver progenitors arise. (C) E9.5 mouse embryo with hepatic bud outlined. (D) Schematic of hepatic bud and adjacent mesoderm-derived populations such as septum transversum mesenchyme (i.e., STM) and sinus venosus (i.e., SV). (E) E13.5 mouse liver and schematic of rudimentary intrahepatic bile duct system in process of being formed through bipotential hepatoblast cell fate decisions into hepatocytes or cholangiocytes.
Once the hepatic diverticulum/bud is formed, further proliferation and expansion of the hepatoblasts commences. In fact, the liver amasses the size of an E9.5 embryo by E13.5 (Figure 1.1). Not until around this time, E11–E14 in mice and 7–10 weeks of gestation in humans, does a subpopulation of hepatoblasts begin to make a decision to enter into the cholangiocyte differentiation program versus the hepatocyte program. In contrast to the other organs derived from the ventral foregut endoderm wherein the ductal systems branch and grow with the organ, the liver forms a branched intrahepatic bile duct network within the generated mass through a multistep process including specification of cholangiocytes and subsequent morphogenesis of the specified cholangiocytes into a tube [Reference Huppert and Iwafuchi-Doi4, Reference Lemaigre5]. This alternative process of tubulogenesis could theoretically bestow the potential of the liver to continually generate a connected intrahepatic bile duct system coordinated with a normal enlarging parenchymal mass and enable the unique ability of the liver to regenerate after injury [Reference Schaub6].
Hepatic Specification Requires Signals from Adjacent Mesoderm-Derived Tissues
Careful orchestration of signals between epithelial, mesenchymal and endothelial cells is required to guide hepatogenesis. The hepatic diverticulum, discernible at approximately 3–4 weeks post human conception and mouse E8.5, is initially composed of columnar epithelium that transitions to a single-layer of pseudostratified epithelium attached to a laminin containing basement membrane [Reference Vestentoft7] (Figure 1.2). This transition is concomitant with interkinetic nuclear migration (INM) wherein nuclei undergo S phase in the basal region and then migrate to the lumenal apical region during mitosis. Circa human gestation day 25 and mouse E9.5, the basement membrane is broken down and the hepatoblasts migrate as cords into the mesenchyme of the septum transversum (STM) intermingling with endothelial cells [Reference Tanimizu and Miyajima8] (Figure 1.2).

Figure 1.2 (A) Schematic of steps during hepatic diverticulum expansion. (B) Laminin immunostaining of E9.5 mouse embryo transverse section through the hepatic bud region Highlighting the breakdown of the extracellular matrix only in the hepatic bud region, allowing migration of hepatoblasts into the STM and surrounding endothelial cells.
Hepatoblasts, are distinguishable from the remainder of the endoderm by the expression of liver-specific genes such as alpha-fetoprotein and albumin, starting at around 7–8 somite stage (mouse E8.5) [Reference Deutsch9]. The thickened epithelium of the VMEL and lateral domains in the closing ventral foregut endoderm are visible morphologically just prior to the onset of liver-specific gene expression. At this time, there are no known liver-specific molecular markers associated with the initial ventral foregut endoderm thickening [Reference Costa10]. However, besides alpha-fetoprotein and albumin, HEX, a divergent homeobox gene, is the earliest gene known to be expressed and required for hepatic development [Reference Bort11]. At mouse E9.5, embryos deficient in HEX have formed the thickening of the ventral foregut endoderm domains, but no outgrowth or expansion of cells positive for liver-specific expressed genes is observed. The primary defect in mouse embryos deficient in HEX appears to be the transition of columnar to a pseudostratified epithelium [Reference Bort11]. Therefore, HEX is required cell-autonomously to drive INM independent of initiating liver-specific gene expression. Similarly, mouse embryos deficient in PROX1, a divergent homeobox gene, exhibit a smaller liver and an absence of hepatoblast migration [Reference Sosa-Pineda, Wigle and Oliver12]. However, in contrast to a deficiency in HEX, the primary defect appears to be due to an increase of E-cadherin expression and a failure to break down the laminin-rich membrane surrounding the hepatic bud. Additional transcription factors including TBX3, HNF6/ONCECUT1, and ONECUT2 play roles in hepatoblast delamination, migration, and expansion suggesting a gene regulatory network [Reference Gerard, Tys and Lemaigre13]. However, the genetic hierarchy still remains to be determined.
The mesoderm-derived STM, sinus venosus and endothelial cells secrete and provide several growth factors, including fibroblast growth factor (FGF), bone morphogenetic protein (BMP), hepatocyte growth factor (HGF) and wingless-related integration site (WNT), that non-cell-autonomously promote hepatoblast proliferation, migration and survival. FGF and BMP emanating from the cardiac mesoderm and the STM, respectively, are further proposed to be directly required to induce liver-specific gene expression within the ventral foregut endoderm.
Fibroblast Growth Factor Induction
Initial studies using ventral foregut endoderm and adjacent mesoderm explanted from mouse embryos prior to initiation of liver-specific gene expression still retained the ability to express liver-specific genes, such as albumin or alpha-fetoprotein, after 48 hours in culture. In contrast, in the absence of mesoderm, explanted ventral foregut endoderm does not initiate expression of liver-specific genes, thereby indicating that liver-specific gene expression requires mesoderm supplied factors. Addition of either FGF1 or FGF2 to ventral foregut endoderm explants without the adjacent mesoderm is sufficient to induce expression of the liver-specific genes alpha-fetoprotein and albumin [Reference Deutsch9]. However, FGF1 and FGF2 double knockout embryos are viable and fertile, demonstrating that both FGF ligands are sufficient but not required to initiate liver-specific gene expression or mediate normal hepatic function [Reference Wang14].
To further refine the requirement of FGF signaling during hepatogenesis and avoid the complicated diversity of FGF ligands that are expressed in the mesoderm adjacent to the ventral foregut endoderm, a multi-targeted receptor tyrosine kinase (RTK) inhibitor (SU5402) was used to abolish all FGF-mediated signaling. Whole mouse embryos prior to initiation of liver-specific gene expression were cultured in the presence or absence of the RTK inhibitor until just before hepatoblast delamination and migration. Interestingly, the anterior part of the hepatic bud in the presence of the inhibitor was thin and lacked an anterior evagination from the closed gut tube while the posterior portion of the hepatic bud was morphologically normal [Reference Wang14]. In the presence of the inhibitor, the liver-specific gene alpha-fetoprotein is expressed in a gradient, low in anterior to a more typical level in the posterior, instead of a uniform level across the whole hepatic bud region [Reference Wang14]. Additionally, HNF4, a transcription factor involved in hepatocyte development [Reference Parviz15], and PROX1 expression were both very weak in the anterior and relatively normal in the posterior hepatic bud region [Reference Wang14]. In support of the RTK inhibitor studies, the presence of a mitogen-activated protein kinase (MEK1/2) inhibitor (U0126) exhibited very similar results, demonstrating a required anterior-posterior gradient of FGF-mediated expression of liver-specific genes and hepatogenesis in whole embryo cultures [Reference Wang14]. Moreover, inhibition of FGF signaling post-coalescence of the two lateral and VMEL domains does not cause a defect in hepatic bud size and morphology [Reference Wang14]. This demonstrates that FGF signaling is required spatially in an anterior-posterior gradient for initiating hepatogenesis and liver-specific gene expression, and temporally in the hepatic specification window during which liver-specific genes are induced (Figure 1.3).
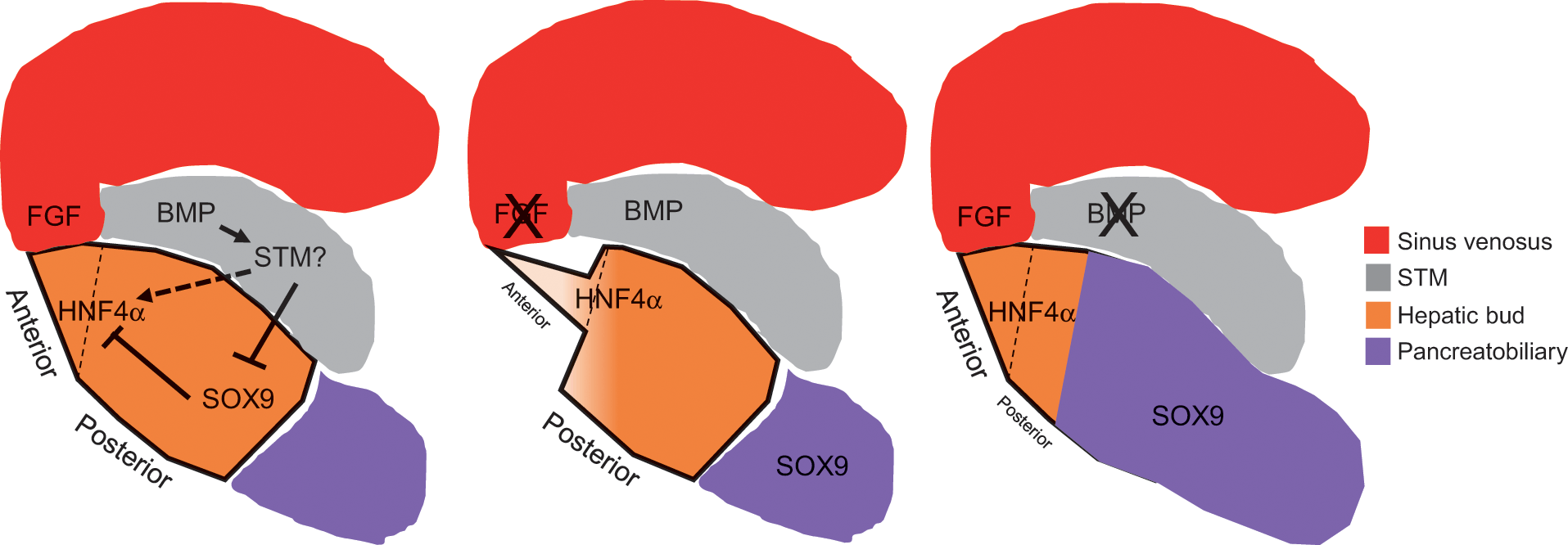
Figure 1.3 Hepatic fate is specified by combinatorial signaling through a progressive series of reciprocal tissue interactions between the ventral foregut endoderm and nearby mesoderm-derived tissues during morphogenetic movements of foregut closure. FGF from the sinus venosus/cardiac mesoderm and BMP from septum transversum mesenchyme cells coordinately specify the hepatic fate in the ventral foregut endoderm and suppress the pancreatic program.
Bone Morphogenetic Protein Induction
Based on this information, an interesting hypothesis is that the anterior hepatic bud requires FGF signaling for its initiation, while the posterior hepatic bud requires an independent signal. An obvious candidate for this signal is BMP. While a role for BMP signaling during hepatogenesis has been established using ventral foregut endoderm and adjacent mesoderm explants from mouse embryos prior to initiation of liver-specific gene expression, a complete understanding is impeded by the redundancy of the pathway components, the requirement for this pathway in multiple earlier developmental processes, and spatial cues that might not be preserved in explants [Reference Wandzioch and Zaret16]. For example, only a subset of BMP4 null embryos are delayed in the formation of the hepatic diverticulum [Reference Rossi17], and BMP4 is also known to be required for establishing mesoderm derivatives that are adjacent to the forming hepatic diverticulum. Therefore, redundancy of alternative BMP ligands may be partially compensating for the BMP4 loss, thereby alleviating hepatic bud defects. Additionally, the hepatic developmental delay observed in BMP4 null embryos may be secondary to an earlier requirement in mesoderm formation.
To further understand the role of BMP during hepatogenesis, whole mouse embryos prior to initiation of liver-specific gene expression were cultured in the presence or absence of a BMP type I receptor antagonist (DMH1), to prevent the phosphorylation of Smad1/5/8, until just before hepatoblast delamination and migration. This experimental approach, which alleviated redundancy, allowed temporal control and spatial morphogenesis to be maintained, and uncovered that BMP signaling has a predominate role in the posterior versus the anterior part of the hepatic bud [Reference Palaria18]. The number of HNF4a-positive hepatoblasts was reduced in the posterior hepatic bud portion lined by the STM versus the anterior hepatic bud portion lined by the sinus venosus, but the overall size of the “bud” was not impacted [Reference Palaria18]. If BMP type I receptor inhibition is performed post-coalescence of the two lateral and VMEL domains, a defect in hepatic bud size, morphology and hepatoblast identity is not observed [Reference Palaria18]. Taken together, these results suggest that BMP signaling is required for initiating liver-specific gene expression for formation of the posterior portion of the hepatic bud (Figure 1.3).
Because the “bud” size was not influenced by inhibition of BMP signaling prior to initiation of liver-specific gene expression, the question remains as to the identity of the cells that contribute to the putative posterior hepatic bud region. Inhibition of BMP type I receptor prior to initiation of liver-specific gene expression results in a reduction of alpha-fetoprotein, but no change in cell death or proliferation of the remaining specified HNF4α-positive hepatoblasts [Reference Palaria18]. Because the general markers of endoderm in the early embryo, such as FOXA1 and FOXA2, are present in the putative posterior hepatic bud region, these results suggest that the endoderm fated cells normally contributing to the hepatoblast population are present but not directed toward the hepatic lineage. Indeed, expression of the posterior foregut endoderm marker PDX1 is shifted anteriorly in whole embryos treated with DMH1 prior to initiation of liver-specific gene expression [Reference Palaria18]. Additionally, the expression of the early pancreatobiliary marker SOX17 and SOX9 transcription factors are expanded from the posterior into the anterior foregut region [Reference Palaria18], uncovering that BMP plays a role in demarcating the hepato-pancreatobiliary boundary (Figure 1.3).
If BMP signals are directly required for hepatic specification, then electroporation of dominant negative forms of BMP type I receptors, dnALK3 and dnALK2, into endoderm prior to initiation of liver-specific gene expression would be predicted to preclude induction of HNF4a. However, similar to controls, dnALK3 or dnALK2 expressing cells in the hepatic bud are also expressing HNF4a [Reference Palaria18]. These results further support the hypothesis that BMP signals are not directly required in the ventral foregut endoderm for initiation of liver-specific gene expression and hepatoblast specification and invoke an additional molecular factor in the STM (Figure 1.3).
To understand how inhibition of BMP signaling influences liver-specific gene expression, the role of SOX9 ectopic expression in the anterior foregut and putative hepatic domain was investigated further. SOX9 is known to negatively regulate the intestinal transcription factor caudal type homeobox2 (CDX2) at the pancreato-duodenal boundary, restricting the expansion of the duodenum into the pancreatic domain [Reference Shih19]. If SOX9 directly influences hepatic specification, then electroporation of SOX9 into the ventral foregut endoderm prior to initiation of liver-specific gene expression would preclude induction of HNF4a. As predicted, forced expression of SOX9 inhibited expression of HNF4a [Reference Palaria18]. Even though SOX9 was sufficient to inhibit HNF4α expression, SOX9 alone was not sufficient to induce pancreatobiliary genes such as PDX1 and SOX17 [Reference Palaria18]. Together, these results demonstrate that SOX9 cannot single-handedly promote pancreatic or biliary fate in the foregut endoderm (Figure 1.3).
Finally, using small molecules to simultaneously inhibit the BMP (DMH1) and FGF (SU5402) signaling pathways results in almost complete loss of the presumptive hepatic bud. HNF4a expression is replaced by ectopic SOX9 expression throughout what is normally the hepatic patterned foregut endoderm [Reference Palaria18]. Therefore, BMP and FGF signaling act from the adjacent STM and sinus venosus mesoderm on complementary regions of the ventral foregut endoderm to pattern and initiate liver-specific gene expression.
Endothelial Cells as an Inductive Signaling Source
After ventral foregut closure through coalescence of the three hepatic domains, the hepatoblasts proliferate and migrate into the surrounding mesenchyme, interacting to form a hepatic bud diverging from the endoderm that becomes vascularized. Loss-of-function studies in mice have demonstrated that kinase insert domain receptor (KDR), which encodes the vascular endothelial growth factor receptor 2 (VEGFR2), is required for hepatic outgrowth, but not hepatic specification [Reference Matsumoto20]. Because of the early embryonic lethality observed in homozygous KDR mutants, an embryonic explant culture system was used to assess the expansion of the albumin-positive hepatic cells. The hepatic bud was isolated at E9.5 and put into culture for 72 hours. The outgrowth of the hepatic endoderm was specifically affected in homozygous KDR mutants, in contrast to the growth of the surrounding fibroblast cells or the initial expression of liver-specific gene expression in the endoderm. Additionally, the hepatic outgrowth observed in the explant cultures, induced by the remnant endothelial cells contained in the wild-type explants, suggests that an intact vasculature and embryo are not necessary for hepatic outgrowth [Reference Matsumoto20].
Single-cell RNA sequencing of three-dimensional hepatic bud organoids reconstituted from human iPSC-derived hepatic endoderm, mesenchymal stem cells and endothelial cells established that the transcriptome single cell-states of the hepatic bud organoids more closely resemble the single-cell transcriptomes of human fetal hepatic cells isolated from samples at gestation weeks 10.5 and 17.5 than human adult liver [Reference Camp21]. Focusing on the hepatic endoderm and endothelial cell interaction, KDR/VEGFR2 inhibitor treatment of human hepatic bud organoids or mouse embryonic stem cell-derived hepatic endoderm co-cultured with endothelial cells resulted in impaired endothelial sprouting and hepatic differentiation [Reference Camp21, Reference Han22], independent of hepatic bud organoid self-condensation driven by mesenchymal cells [Reference Camp21]. As expected, since the majority of hepatic endoderm does not express KDR/VEGFR2, the transcriptome of cultures consisting solely of human iPSC-derived hepatic endoderm treated with KDR/VEGFR2 inhibitors were not changed [Reference Camp21]. Because cultures consisting of only human iPSC-derived hepatic endoderm do express some mature hepatocyte markers, but their overall transcriptome is more similar to mouse E8.5–E10 liver, the conclusion that the endoderm-endothelial interaction is not required for initial hepatic specification is further supported [Reference Camp21].
Differentiation of Hepatoblasts into Hepatocytes and Cholangiocytes
Hepatoblast Potential
All previous human gene expression studies, culture experiments, as well as transgenic mouse approaches support the view that hepatocytes and cholangiocytes are derived from hepatoblasts, the hepatic progenitor [Reference Huppert and Iwafuchi-Doi4, Reference Lemaigre5]. However, a knowledge gap remains as to whether (1) a single hepatoblast is truly bipotential and has the capability in vivo to give rise to both hepatocytes and cholangiocytes, (2) there are subpopulations of unipotent hepatoblasts that produce only hepatocytes or cholangiocytes, or (3) all hepatoblasts are equipotential and the fate of their progeny is dependent on their location as the hepatic architecture is established. Using single cell labeling approaches to fate map the cellular contribution of individual hepatoblasts supports the concept that single hepatoblasts exist that are bipotential.
In order to map the potential of hepatoblasts, transgenic mouse models expressing a tamoxifen-inducible Cre recombinase under control of the endogenous FOXA2 promoter were used to activate expression of a Cre-mediated lacZ reporter (ROSA26 Reporter – lacZ). Thereby enabling permanent labeling of single FoxA2-positive unspecified endoderm cells prior to liver-specific gene expression and tracking the progeny at E16.5, when the gut tube has formed, the endoderm organs have specified, and cell types within each endoderm-derived organ are differentiating. To identify whether the clonal descendants of a single FOXA2-positive endoderm cell had become a hepatocyte and/or cholangiocyte, the cell markers HNF4a or SOX9 were used to identify hepatocytes or cholangiocytes, respectively. Combining these cell identity markers with the lacZ fate tracing label explicitly demonstrated that clonal descendants originating from a single FOXA2-positive endoderm cell have the potential to give rise to both hepatocytes and cholangiocytes [Reference El Sebae1].
To further support the conclusion that hepatoblasts are bipotential, a similar cell labeling approach was used to fate map the cellular contribution of leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5)-positive cells. LGR4 and LGR5 paralog receptors and their cognate R-spondin ligands potentiate Wnt/β-catenin signaling and promote tissue homeostasis through proliferation of epithelial stem cell compartments [Reference Planas-Paz23]. Expression of LGR5 in the adult homeostatic liver is restricted to pericentral hepatocytes [Reference Planas-Paz23], but is upregulated in response to liver damage [Reference Huch24]. R-spondin, LGR4 and LGR5 controls metabolic liver zonation and is a hepatic size rheostat during development, homeostasis and regeneration [Reference Planas-Paz23]. To alleviate the caveat of labeled cells becoming dispersed from clones, a Cre-mediated multicolor fluorescent confetti reporter (ROSA26 reporter – Confetti) was used to irreversibly label the progeny of cells with mutually exclusive fluorescent proteins. Scoring was performed on individual clones of single colors adjacent to portal tracts where hepatocytes and cholangiocytes are juxtaposed. Labeling LGR5-positive hepatoblasts at E9.5 when the hepatic bud has formed reveals clones near portal tracts in postnatal liver (P0-P17) that contain hepatocytes and cholangiocytes (bipotential – 50%) or hepatocyte only (unipotential) [Reference Prior25]. No unipotential cholangiocyte clones were observed [Reference Prior25]. Therefore, fate tracing individual Lgr5-positive hepatoblasts at E9.5 demonstrates that a subset of hepatoblasts is bipotent. In contrast, labeling Lgr5-positive hepatoblasts at E13.5 results in only hepatocyte clones, indicating that LGR5-postive hepatoblasts after E9.5 are unipotential and committed to hepatocyte fate [Reference Prior25]. Additionally, labeling LGR5-positive cells at E7.5 and E8.5, prior to initiation of liver-specific gene expression, does not label hepatic progeny in the postnatal liver [Reference Prior25], all indicating that LGR5 does not mark the ventral foregut endoderm cells before hepatic specification. These results suggest that LGR5-positive cells are heterogeneous in their potential and lose their bipotency during developmental progression.
Hepatoblast Transcriptomic Signatures at Single-Cell Resolution
Understanding the mechanisms that determine how cells activate and repress genes in the correct spatial and temporal pattern to generate different cell types, further regulate cell type differentiation, and maintain cell identity is a key question in biology. Cell fate decisions during lineage differentiation are coordinately regulated at the transcriptional level by the activation and repression of specific gene sets. How and when individual bipotential hepatoblasts differentiate into hepatocytes and cholangiocytes remains unclear.
The transcription factors TBX3, CEBPA, PROX1, and HNF4a have been shown, through loss-of-function experiments, to promote hepatocyte differentiation beginning at the time of hepatic specification [Reference Gerard, Tys and Lemaigre13], whereas the differentiation of cholangiocytes is proposed to begin before E15.5 as a result of the transcription factors HNF6/ONECUT1, SOX4, SOX9, and HNF1b promoting cholangiocyte differentiation [Reference Gerard, Tys and Lemaigre13]. Despite these findings, the timing and mechanisms underlying the differentiation of individual hepatoblasts into hepatocytes and cholangiocytes remain unclear.
Single-cell RNA sequencing can be used to overcome the lack of cellular markers that can be used to efficiently distinguish and purify subpopulations of hepatoblasts, hepatocytes, and cholangiocytes from different stages of the developing fetal liver, facilitating further profiling of molecular differences. This approach enables identification of individual cell states, computational ordering of cells along a developmental trajectory, and construction of gene regulatory networks. Furthermore, single-cell transcriptomic analysis of hepatoblasts, hepatocytes and cholangiocytes can be used to delineate when hepatoblasts begin to change their cell identity state and the amount of heterogeneity within the hepatoblast population.
Six different cell types, including endothelial cells, erythrocytes, hepatoblasts, macrophages, megakaryocytes and mesenchymal cells are discernible by single-cell RNA sequencing at different proportions in fetal mouse liver from E11.5 to E16.5 [Reference Su26]. The proportion of erythrocytes increases from E11.5 to E14.5 and then decreases at E16.5. Mesenchymal cell proportion decreases in a stepwise manner concomitant with an increase in hepatoblast cell number from E11.5 to E16.5 [Reference Su26]. Interestingly, even though a majority of hepatoblasts are thought to be bipotential, only a minority of hepatoblasts co-express hepatocyte- (KRT8 and KRT18) and cholangiocyte-specific markers (KRT7 and KRT19) from E11.5 to E16.5 [Reference Su26].
To circumvent the large number of hematopoietic cells in the fetal liver, delta-like 1 homolog (DLK1), a general hepatoblast cell surface marker that is expressed until the neonatal stages, can be used for fluorescence-activated cell sorting (FACS) and to enrich single-cell RNA sequencing of hepatoblasts [Reference Tanimizu27]. Although differentiation and proliferation are usually inversely correlated during cell differentiation, hepatoblasts aligned in pseudotime of increasing time points (E10.5 to E13.5) are both proliferating and quiescent regardless of developmental age [Reference Yang28]. Therefore, single-cell RNA sequencing analyses suggest that the differentiation state of proliferating and quiescent hepatoblasts are synchronized during early liver development with the proliferation rate steadily decreasing with age.
Hierarchical cluster analysis of genes expressed in single hepatoblasts from E10.5 to E13.5 identified 383 genes whose expression levels increased gradually with age [Reference Yang28] without initiating expression of cholangiocyte genes from E11.5 to E16.5 [Reference Su26]. Gene ontological analyses show enrichment of categories associated with hepatocyte metabolic, detoxification and transport function [Reference Su26, Reference Yang28]. Some differences between hepatoblasts and hepatocytes include low or no expression of hepatic progenitor markers, such as GPC3, DLK1 and AFP, but higher expression of hepatocyte genes, such as FBP1, MAT1A, and SULT1A1 [Reference Su26]. Based on these data, hepatoblasts enter the hepatocyte transcriptional program after hepatic specification from endodermal progenitors and gradually move in unison toward the hepatocyte fate.
Because the differentiation cell state of individual hepatoblasts appears to be synchronized at each age, bulk RNA sequencing of DLK1-positive hepatoblasts can be performed to develop a robust gene expression signature at each age from E10.5 to E18.5. Hierarchical clustering of gene expression signatures identifies 4,077 variably expressed genes with strong correlation with single-cell RNA sequencing data during hepatoblast to hepatocyte differentiation [Reference Yang28]. The differentially expressed genes form two prominent clusters revealing hepatoblast to hepatocyte differentiation begins at E13.5 through a switch of a gene signature that is down-regulated in one cluster and up-regulated in the other. The transcriptomic signature suggests that the hepatocyte cell identity seems to be locked in by E15.5 [Reference Yang28]. The synchronicity and progressive linear differentiation suggest that the hepatocyte lineage specification is the default cell identity.
To obtain insights into cholangiocyte specification and differentiation, the dynamics of gene expression in cells predicted to be in the cholangiocyte program were analyzed from E11.5 to E17.5. Hierarchical clustering of gene expression established three groups, indicating that predicted cholangiocytes identified at E11.5 and E12.5 are in a different cluster from predicted cholangiocytes at later time points. Gene ontological analyses exhibited enrichment of categories associated with hepatocyte metabolic, detoxification and transport functions [Reference Yang28]. From E13.5 to E17.5, gene ontological analyses exhibited enrichment of cell adhesion, migration, and tube morphogenesis [Reference Yang28], suggesting that newly specified cholangiocytes retain transcriptomic features of their hepatoblast progenitors, whereas cholangiocyte maturation involves inducing the expression of genes required for their structure and function. However, the predicted cholangiocytes at later time points have gene signatures that are heterogeneous and not age dependent. Therefore, as individual cholangiocytes begin to be specified and diverge from the default hepatoblast to hepatocyte path, they upregulate a cholangiocyte transcriptional program coincident with repression of the hepatocyte transcriptional program.
In contrast to hepatoblast to hepatocyte differentiation, the percentage of proliferating cells increased in the hepatoblast to cholangiocyte differentiation groups based on hierarchical clustering of gene expression. KRT19 is rarely detected in cells predicted to be cholangiocytes from E11.5 to E12.5. However, cells that were KRT19-positive displayed a significant increase of proliferating cells [Reference Yang28]. Interestingly, a quiescent state is proposed to be important for initial hepatoblast to cholangiocyte specification due to the observation that homozygous TBX3-deficient mouse embryos have reduced hepatoblast proliferation coincident with predominant cholangiocyte instead of hepatocyte differentiation [Reference Ludtke29]. Thus, hepatoblast to cholangiocyte differentiation involves mechanisms of coordinating proliferation and differentiation distinct from hepatoblast to hepatocyte differentiation. Additionally, given the heterogeneity in the transcriptional signatures observed at the same developmental stage, cholangiocyte differentiation takes a more nonsynchronous differentiation path compared to hepatocyte differentiation [Reference Yang28].
The question regarding whether a hepatobiliary progenitor population present in fetal liver is maintained in adult normal liver is still debated. Hepatic cells reported to be EPCAM- and NCAM-positive are found to reside as a subset of the cholangiocyte population in the fetal liver and to be retained in the canals of Hering at the interface of hepatocyte bile canaliculi and cholangiocyte constructed intrahepatic bile ducts of normal adult liver [Reference Schmelzer30]. To determine the transcriptional signature of these cells and whether they maintain a hybrid hepatocyte and cholangiocyte transcriptional signature, single-cell RNA sequencing was performed on human fetal (second trimester, 15–21 post conceptual week) and adult human hepatic cells. FACS was used to negatively select red blood cells (CD235a) and immune cells (CD45), and positively select EPCAM and NCAM to enrich for potential hepatic progenitors and compare to EPCAM-negative and asialoglycoprotein receptor 1 (ASGPR1)-positive hepatocytes [Reference Segal31]. Transcriptionally, the potential progenitors co-express genes associated with both hepatocyte and cholangiocyte lineage, but with an expression profile that can be distinguished from fetal hepatocytes and mature cholangiocytes. Genes, including TACSTD2, CLDN4, CLDN10, and KRT7 are enriched in adult potential progenitors whereas CLDN6, STAT4, MCAM, CDH6, and STAT1 are enriched in fetal potential progenitors [Reference Segal31]. A subset of human adult cholangiocytes has been verified in multiple studies to express both hepatocyte and cholangiocyte genes [Reference Aizarani32, Reference MacParland33]. These results suggest that there is molecular heterogeneity within the EPCAM-positive population similar to the nonsynchronous cholangiocyte differentiation observed in mice [Reference Yang28].
The human potential progenitor transcriptional signature has been compared to mouse chronic injury models. In such models hepatocyte-derived proliferative cholangiocytes have a signature more similar to mouse hepatocyte-derived proliferative cholangiocytes [Reference Tarlow34]. The overlapping signature includes AHSG, RBP4, SFRP5 and MCAM, while the mature cholangiocyte markers KRT7, MUC1, TSPAN8 and TFF2 are downregulated [Reference Segal31]. Although mouse studies using genetic reporters to trace the hepatocyte or cholangiocyte lineages reveal no evidence of contribution from a stem or progenitor cell population held in reserve from development for liver regeneration, the ability of the potential human hepatic progenitors to contribute to a regenerative response is unknown. Additionally, without labeling and fate tracing the potential progenitor population, the transient or permanent transcriptional cell state of the identified cell population remains to be discovered during homeostasis, injury and disease conditions. The possibility remains that the defined progenitor transcriptional cell state could involve differentiated hepatocytes and cholangiocytes stochastically cycling at any given time versus potential progenitor cells held in reserve from the fetal liver.
Intrahepatic Bile Duct Development
Cholangiocyte differentiation follows a path that diverges from the linear continuum of hepatoblast to hepatocyte differentiation in a nonsynchronous manner. Cholangiocyte identity is initiated specifically in the periportal region where hepatoblasts entering into the transcriptionally active cholangiocyte program form a structure termed the ductal plate (Figure 1.4). This implies that hepatoblasts are not pre-fated to the cholangiocyte lineage but rather their location and signals arising from a niche influence their ultimate fate [Reference Huppert and Iwafuchi-Doi4, Reference Lemaigre5].
Figure 1.4 Model for intrahepatic bile duct formation. (A) Hepatoblasts that are in close association with the portal vein mesenchyme activate the cholangiocyte transcriptional program, including SOX9, and repress the hepatocyte program, including HNF4α. Next, primitive ductal structures (PDSs) or lumenal structures composed of asymmetrical gene-expressing cells form. These lumenal spaces are surrounded by sox9+ hnf4α− tgfbr2− cells on the portal vein side and Sox9− Hnf4α+ Tgfbr2+ cells on the parenchymal side. Symmetrical gene-expressing cells encircle the lumenal ductal structures of mature ducts. The remaining ductal plate cells that are not incorporated into an intrahepatic bile duct regress back to hepatocyte-like cells. (B) Cholangiocytes are specified in the region adjacent to the portal vein system forming a ductal plate in a proximal to distal direction. The specified cholangiocytes are quickly incorporated from the ductal plate into a dense homogeneous network of tubes. The homogeneous network begins to reorganize into a hierarchical network between mouse E17 and E18 as hepatocytes begin secreting bile. (C) Three-dimensional association of the portal vein system (white) and the intrahepatic bile duct system (black) visualized by retrograde ink injections into the left lobe of the mouse liver.
Intrahepatic Bile Duct Morphogenesis
Around E11–E14 in mice and 7–10 weeks of gestation in humans, a subpopulation of hepatoblasts forms a temporary structure appearing as a band of potential cholangiocytes, termed the ductal plate, encircling the portal veins [Reference Vestentoft7, Reference Antoniou35] (Figure 1.4). Remodeling of the ductal plate or tubulogenesis begins around E15–E17 in mice and 11–15 gestational weeks in humans [Reference Vestentoft7, Reference Antoniou35] (Figure 1.4). Transient asymmetrical primitive ductal structures (PDSs) are observed in mice and humans [Reference Raynaud36]. The PDS is asymmetrical; cells on the portal vein side of the lumen express the marker SOX9, compared to cells on the parenchymal side that express HNF4a and TGFBR2. A mature duct is symmetrical with all cells expressing SOX9 [Reference Antoniou35, Reference Raynaud36]. Detailed analysis using immunostaining suggests that there is a radial progression of differentiation – mature cells on the portal vein side of the lumen promote differentiation of the neighboring less mature cells on the parenchymal side of the lumen [Reference Antoniou35], perhaps through ERM-dependent apical constriction involving NF2 [Reference Benhamouche-Trouillet37]. However, it remains unclear if cells from the ductal plate move and contribute to the less mature cells of the forming lumen or more hepatoblasts are recruited to contribute to the lumen. Given the expression of HNF4a in the less mature cells on the parenchymal side of the PDS, the more plausible explanation is that hepatoblasts are recruited to contribute to the forming lumen.
Remodeling of specified cholangiocytes into intrahepatic bile ducts starts at the larger hilar/proximal portal vein regions and moves toward the peripheral/distal region of the liver following the portal system. Three-dimensional analyses indicate that newly committed cholangiocytes are specified peripherally and are quickly incorporated into a homogeneous network encircling the portal vein [Reference Vestentoft7, Reference Takashima38, Reference Tanimizu39] (Figure 1.4). In mice, the homogenous network is rearranged into a hierarchical network between E17 and E18 (Figure 1.4), the timing of which correlates with lengthening of the bile canalicular network, hepatocyte excretion of bilirubin into bile, and the presence of bile in the intestine [Reference Tanimizu39]. If efflux of bile from the hepatocyte canalicular membrane is blocked during this critical period, the structural rearrangement and formation of the intrahepatic bile duct hierarchical network is inhibited [Reference Tanimizu39]. Whether the lumenal enlargement observed in the hierarchical network occurs through localized proliferation of cholangiocytes, movement and incorporation of nearby cholangiocytes, or stretching and enlargement of cholangiocyte cell size remains to be experimentally determined.
The intrahepatic bile duct architecture remains incomplete in the liver periphery of mice until 1 week of age and humans during the first years of life [Reference Tanimizu39, Reference Van Eyken40]. Therefore, there is progressive assembly of the intrahepatic bile duct architecture coincident with the enlargement of the liver during normal growth in childhood. The final intrahepatic bile duct hierarchical architecture consists of large ducts running along portal veins and small channels forming a mesh-like network around portal veins (Figure 1.4).
Transforming Growth Factor Beta Signaling
Given that cholangiocyte specification and tubulogenesis take place in the vicinity of the portal vein, local cues must be involved. The mRNA of both TGFB2 and TGFB3 ligands are predominantly expressed by portal vein myofibroblasts, while TGFB1 is produced throughout the hepatic parenchyma [Reference Antoniou35]. A portal to central gradient of TGFB activity is observed at E12.5 using a mouse transgenic reporter [Reference Clotman41]. These data suggest that spatial activation of the TGFB pathway in the region of the portal vein may induce cholangiocyte specification and thereby ductal plate formation. The dynamic expression pattern of TGFBR2 suggests that the TGFB pathway also plays a role in tubulogenesis. In support of this, immunostaining of TGFB2 and TGFB3 demonstrates asymmetrical localization specifically to the parenchymal side of the PDS coincident with localization of TGFBR2 [Reference Antoniou35]. At the onset of cholangiocyte specification, TGFBR2 and ligands are expressed in all cells of the ductal plate. In mature ducts, all cholangiocytes are negative for TGFBR2 expression [Reference Antoniou35]. This model suggests that TGFB activity transiently induces cholangiocyte specification, PDS, and then is downregulated.
Several functional studies support the premise that TGFB signaling plays a role in cholangiocyte specification. Mouse E12.5 fetal liver explants or E14.5 hepatoblast cell line exposed to TGFB ligands reduce the mRNA expression of hepatocyte markers and increase the mRNA expression of cholangiocyte markers [Reference Antoniou35, Reference Clotman41]. Additionally, human embryonic stem cell generated hepatoblasts, treated with TGFBR2, TGFB1, or TGFB2, but not TGFB3, increases mRNA expression of cholangiocyte markers and decreases mRNA expression of hepatocyte markers in vitro [Reference Takayama42]. All supporting the ability of the TGFB pathway to induce cholangiocyte specification in the context of functional Notch signaling. However, a requirement for TGFB signaling in vivo is unclear. Inhibiting TGFB activity by anti-TGFB neutralizing antibody at E10.5 shows a qualitative reduction in the cholangiocyte KRT-positive cells around the portal vein [Reference Clotman41]. However, deletion of TGFBR2 specifically in hepatoblasts does not result in an intrahepatic bile duct phenotype [Reference Schaub6]. Therefore, the TGFB pathway may only be required temporally at the very beginning of cholangiocyte specification, either in the process of setting up competency or direct regulation of cholangiocyte target genes.
Two different transcription factor families are proposed to impact TGFB activity. CEBPA and CEBPB negatively and positively regulate TGFBR2, respectively [Reference Takayama42]. Both CEBP family members are recruited to the same DNA-binding site in the TGFBR2 promoter region in cells differentiated to the hepatocyte-like or cholangiocyte-like fate from human embryonic stem cells [Reference Takayama42]. This suggests that the ratio of CEBPB to CEBPA governs the hepatoblast cell fate decision by regulating the expression of TGFBR2. Additionally, the ONECUT family of transcription factors, HNF6/ONECUT1 and ONECUT2 modulate the level of TGFB activity. Loss of HNF6/ONECUT1 and/or ONECUT2 results in ectopic TGFB activity allowing formation of primitive ductules within the parenchyma instead of restricting cholangiocyte differentiation to the portal vein region [Reference Clotman41]. However, no evidence of HNF6/ONECUT1 directly binding to the TGFBR2 promoter has been found [Reference Takayama42]. These findings allude to indirect regulation of TGFB by the ONECUT family and imply a feedback loop involving the CEBP family of transcription factors.
Notch Signaling
Clinically, mutations in the NOTCH pathway manifest as Alagille syndrome (see Chapter 14), which is a multisystem autosomal dominant disorder. Clinical features of Alagille syndrome are wide-ranging, and include hepatic (e.g., ductopenia) and extra-hepatic (e.g., facial, cardiovascular, skeletal, ocular) manifestations. Mutations in JAG1 or, to a lesser extent, NOTCH2 account for >95% of cases [Reference McDaniell43, Reference Warthen44]. The multisystem effects of Notch signaling mutations highlight the importance of this pathway in development.
Without question, JAGGED1-mediated NOTCH signaling is essential to intrahepatic bile duct development. While TGFβ ligands are secreted, NOTCH pathway ligands and receptors are integral membrane proteins requiring close cell-cell association between signaling and receiving cells to elicit signal transmission. During hepatic development, periportal mesenchyme expresses JAG1 resulting in NOTCH receptor activation in periportal adjacent hepatoblasts [Reference Huppert and Iwafuchi-Doi4]. Loss of NOTCH signaling in mice, either through haploinsufficiency of JAG1 or deletion of NOTCH2 from hepatoblasts at mid-gestation, results in decreased cholangiocyte specification and stunted intrahepatic bile duct architecture [Reference Jeliazkova45–Reference Thakurdas47]. SOX9, one of the first genes expressed in hepatoblasts entering the cholangiocyte transcriptional program, is a NOTCH target gene [Reference Zong48]. Two cis-regulatory DNA-binding sites with the SOX9 promoter suggest that a specific level of Notch activation may be necessary to induce the correct expression level for cholangiocyte specification.
The HIPPO/YAP pathway has also demonstrated the ability to influence the cholangiocyte fate, but in a Notch-dependent mechanism. Increased YAP activity directly or through NF2 has the potential to induce hepatoblast and hepatocyte conversion to a cholangiocyte-like fate [Reference Yimlamai49, Reference Zhang50]. These phenotypes are very reminiscent of the induced hepatoblast and hepatocyte conversion to cholangiocytes upon constitutive NOTCH activation in mice [Reference Jeliazkova45, Reference Sparks46, Reference Zong48, Reference Tchorz51]. DNA-binding of TEAD4, the transcription factor that mediates YAP-induced gene expression, within the Notch2 promoter region suggests that the phenotype observed upon YAP activation is a NOTCH2-dependent mechanism [Reference Yimlamai49]. Epistatic studies have confirmed this model. Reducing the NOTCH2 signaling activity, through hepatoblast-specific deletion, suppresses the NF2 increased cholangiocyte differentiation phenotype and rescues the stunted intrahepatic bile duct architecture associated with the loss of NOTCH2 [Reference Wu52].
Potential Developmental Underpinnings of Ductal Plate Malformation
Ductal plate malformations (DPMs) are defined as the retention of fetal ductal plate-like structures, such as cholangiocytes not incorporated into a bile duct structure, in the adult as observed in patients with autosomal recessive polycystic kidney disease (ARPKD), congenital hepatic fibrosis, autosomal dominant polycystic kidney disease (ADPKD), Caroli disease, Caroli syndrome, and von Meyenburg complexes (see Chapter 41) [Reference Raynaud36, Reference Desmet53, Reference Huppert54]. DPM may occur due to abnormal cholangiocyte differentiation or tubulogenesis along any stage of intrahepatic bile duct development. Lineage tracing SOX9-positive cells in mice strongly suggest that the remaining unincorporated ductal plate cells default into hepatocytes without apoptosis or proliferation [Reference Carpentier55]. If the unincorporated ductal plate cells do not receive or are unresponsive to the proper signals, they may contribute to DPM. Additionally, defects in signaling pathways (e.g., TGFB or NOTCH) or transcription factors (e.g., SOX9, HNF6/ONECUT1, or HNF1B) can cause defects in PDS formation leading to developmental delays or cholangiocytes not fully committing to the cholangiocyte program [Reference Raynaud36], thereby triggering improper remodeling of the ductal plate, cholangiocyte polarity and defects in tubulogenesis. Thus, there is a high level of coordination that must regulate sequential tubulogenesis and regression of the ductal plates along portal veins within the three-dimensional space of the liver to connect the entire intrahepatic bile duct system to the extrahepatic ductal system. This indicates that careful orchestration of signals between epithelial and mesenchymal cells is required to guide intrahepatic bile duct formation [Reference Huppert and Iwafuchi-Doi4, Reference Lemaigre5, Reference Gerard, Tys and Lemaigre13].