
Advances in X-ray Analysis, Sixteenth Annual Conference on Applications of X-ray Analysis, August 9-11, 1967
Volume
11
-
1967
- This volume was published under a former title. See this journal's title history.
Contents
Other
Foreword
-
- Published online by Cambridge University Press:
- 06 March 2019, p. vi
-
- Article
- Export citation
Preface
-
- Published online by Cambridge University Press:
- 06 March 2019, p. vii
-
- Article
- Export citation
Research Article
Recent Advances in Quantitative X-Ray Spectrometric Analysis by Solution Techniques
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 1-22
-
- Article
- Export citation
X-Ray Fluorescence Spectroscopy in the Analysis of Ores, Minerals, and Waters*
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 23-39
-
- Article
- Export citation
Common Sources of Error in Electron Probe Microanalysis
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 40-55
-
- Article
- Export citation
X-Ray Spectrographs Analysis of Traces in Metals by Preconcentration Techniques
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 56-62
-
- Article
- Export citation
Theoretical Correction for Coexistent Elements in Fluorescent X-Ray Analysis of Alloy Steel
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 63-94
-
- Article
- Export citation
Micro Fluorescent X-Ray Analyzer
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 95-104
-
- Article
- Export citation
Use of Primary Filters in X-Ray Spectrography: A New Method for Trace Analysis*
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 105-113
-
- Article
- Export citation
-
A method to improve the detectability of trace elements by X-ray fluorescent spectrography is described. The method consists of using appropriate filters in the primary, or exciting, beam. The effects of using filters in the primary beam on the peak-to-background ratio R of a fluorescent line have been analyzed on theoretical grounds. In fact,
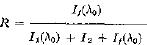
where I1(λ0) is the intensity of the analytic fluorescent line, I1(λ0) is the background intensity due to coherent and Compton scattering of the primary radiation by the specimen, and I2 is the background intensity due to scattering of the fluorescent radiation by the analyzing crystal. Analytical expressions were derived for I1(λ0), I2, and I1(λ0), from which it has been concluded:
1. The ratio I1(λ0)/I1(λ0) decreases when the filter used has its absorption edges at wavelengths longer than λ0.
2. The ratio I2/I1(λ0) can be separated into two parts which vary in opposite ways. The influence of these two parts on the value of R is discussed in the text.
It is then shown that the method should work well at short wavelengths and less well at longer wavelengths. The method was tested in the difficult case where overlapping of the analytical line with a characteristic line of the tube occurred, i.e., in the determination of traces of selenium by using tungsten radiation. The analytic line Se Kα. has a wavelength of 1.106 Å, while W Lγ1occurs at 1.098 Å. There is a marked effect of the filter thickness on the detectability; an optimum thickness appears to exist for each case. In the analysis of selenium, the best filter thickness (which can be selected by mere inspection of the diagrams reproduced in the text) increased the detectability of selenium traces by an order of magnitude. Finally, from statistical considerations, the quantity tσ2 is proposed as an index of the effectiveness of the filter: the smaller tσ2 is, the better the filter is. Here σ is the standard deviation of the intensity of the analytic line and t is the total counting time spent on the measure of the analytic line arid background. In order to study the dependence of the index tσ2 on the filter thickness, measurements were made on samples of sugar containing known concentrations of strontium. Then tσ2 was plotted against the thickness of the filter for each concentration; these curves do show a minimum. Thus, an optimum filter thickness exists in each case.
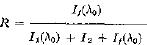
Precision and Accuracy of Silicate Analyses by X-Ray Fluorescence
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 114-128
-
- Article
- Export citation
Applications of Computerized Statistical Techniques in Quantitative X-Ray Analysis
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 129-149
-
- Article
- Export citation
X-Ray Fluorescence Analysis of a Manganese Ore
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 150-157
-
- Article
- Export citation
-
To replace time-consuming chemical analysis, a procedure was developed for applying X-ray fluorescence to the analysis of a manganese ore. This X-ray fluorescence method is rapid; the total time for both sample preparation and analysis is ½ hr. The method is also simple enough for routine laboratory use. The components determined, the concentration ranges, and the agreement between chemical and X-ray analysis in terms of standard deviation are:
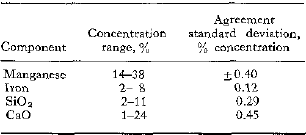
The agreements between chemical and X-ray analysis noted for manganese and iron are obtained by correcting for CaO concentration when determining the manganese and for manganese and SiO2 concentrations when determining the iron. The corrections employ empirical equations developed by a multiple regression technique.
Sample preparation is reduced to a minimum because it consists of only two steps, pulverization of the manganese ore followed by briquet ring. Four minutes of grinding with Boraxo as the grinding aid gives sufficient uniformity to minimize the effect of particle size variation. The ground mix of manganese ore and Boraxo is pressed into 1.25-in. briquettes for analysis.

Total Nondestructive Analysis of Caas Syenite
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 158-163
-
- Article
- Export citation
X-Ray Fluorescence of Suspended Particles in a Liquid Hydrocarbon
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 164-176
-
- Article
- Export citation
The Effect of Surface Roughness in Polymers on X-Ray Fluorescence Intensity Measurements
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 177-184
-
- Article
- Export citation
Production Control of Gold and Rhodium Plating Thickness on Very Small Samples by X-Ray Spectroscopy
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 185-190
-
- Article
- Export citation
A Study of X-Ray Fluorescence Method with Vacuum and Air-Path Spectrographs for the Determination of Film Thicknesses of II-VI Compounds
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 191-203
-
- Article
- Export citation
Rare-Earth Analyses by X-Ray-Excited Optical Fluorescence
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 204-213
-
- Article
- Export citation
Nondispersive X-Ray Fluorescent Spectrometer
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 214-229
-
- Article
- Export citation
The Influence of Sample Self-Absorption on Wavelength Shifts and Shape Changes in the Soft X-Ray Region: The Rare-Earth M Series
-
- Published online by Cambridge University Press:
- 06 March 2019, pp. 230-240
-
- Article
- Export citation